This 2018 Chinese review concerned advanced glycation end products (AGE) mobility interventions:
“Only a limited number of studies have focused on measuring the effects of low AGEs levels or AGEs inhibitors on mobility, although many observational human studies and in vitro studies have reported the correlation of AGEs with and the contribution of AGEs to mobility, particular in diseases such as:
- osteoporosis,
- cartilage degradation,
- osteoarthritis and
- sarcopenia.
There is insufficient information from previous animal and human studies for use as a reference to determine the intervention period. Although serum AGEs levels can be easily affected by a lower AGEs diet or AGEs inhibitors, it may take longer to see the changes in certain organs or tissues, as a result of a reduction in AGEs accumulation.”

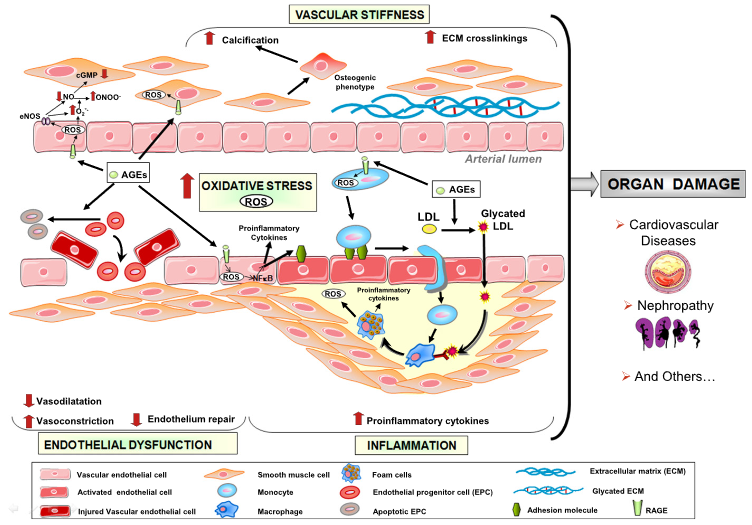
“Effect of AGEs on apoptosis signalling. AP-1, activator protein 1; ERK, extracellular signal-regulated protein kinases; IGF-I, insulin-like growth factor I; IL-6, interleukin-6; JAK, Janus kinase; JNK, c-Jun N-terminal kinases; MEK, mitogen-activated protein kinase; NF-κB, nuclear factor kappa B; p38 MAPK, p38 mitogen-activated protein kinase; RAGE, receptor for AGEs; STAT3, signal transducers and activators of transcription 3; TGF-β, transforming growth factor-β”
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6180645/ “Role of advanced glycation end products in mobility and considerations in possible dietary and nutritional intervention strategies”
Citations aren’t validations of the reference’s quality and strength of evidence. This review would have benefited from not citing reviews that contained misrepresentations, such as one mentioned in Wikipedia is a poor source of information on advanced glycation end products (AGEs).
I came across this review as a result of it citing the excellent 2008 rodent study Oral Glycotoxins Determine the Effects of Calorie Restriction on Oxidant Stress, Age-Related Diseases, and Lifespan which found:
“Higher levels of oxidant AGEs in offspring of Reg-F0 dams may be attributable to placental transmission from mothers with high AGE levels. These high intrauterine AGE levels may predispose the offspring to the development of chronic inflammation and diseases in adulthood, such as insulin resistance and diabetes.
Increasing the intake of AGEs in the diet erases the benefits of CR [calorie restriction]. OS [oxidant stress] can be reduced, and healthspan increased, in mice fed a diet that is restricted in the content of AGEs.
The beneficial effects of a CR diet may be partly related to reduced oxidant intake rather than decreased energy intake.”