Starting this blog’s twelfth year by curating a poorly-done 2026 review of Nrf2 and its capability to change a person’s development of Parkinson’s disease. I’ll emphasize precedent conditions that if not effectively dealt with in youth, can’t prevent PD from occurring at some later life stage.
“This review explicitly examines how age-associated decline in NRF2 responsiveness intersects with redox imbalance, mitochondrial dysfunction, proteostatic failure, and neuroinflammation, core mechanisms shared between aging and PD. PD unfolds through a complex interplay of cellular stress and immune responses. Oxidative stress, mitochondrial dysfunction, and chronic neuroinflammation converge to damage dopaminergic neurons, with microglia playing a central role in amplifying this injury.
NRF2 emerges as a key regulator of antioxidant defenses, inflammatory balance, and mitochondrial protection, offering a promising target for clinical intervention. NRF2 activity favors the anti-inflammatory microglial over the pro-inflammatory phenotype. Decline in NRF2 inducibility with age impairs microglial clearance, promotes neuroinflammation, and reduces antioxidant defenses, while NRF2 activation restores protective functions and offers a promising therapeutic target.
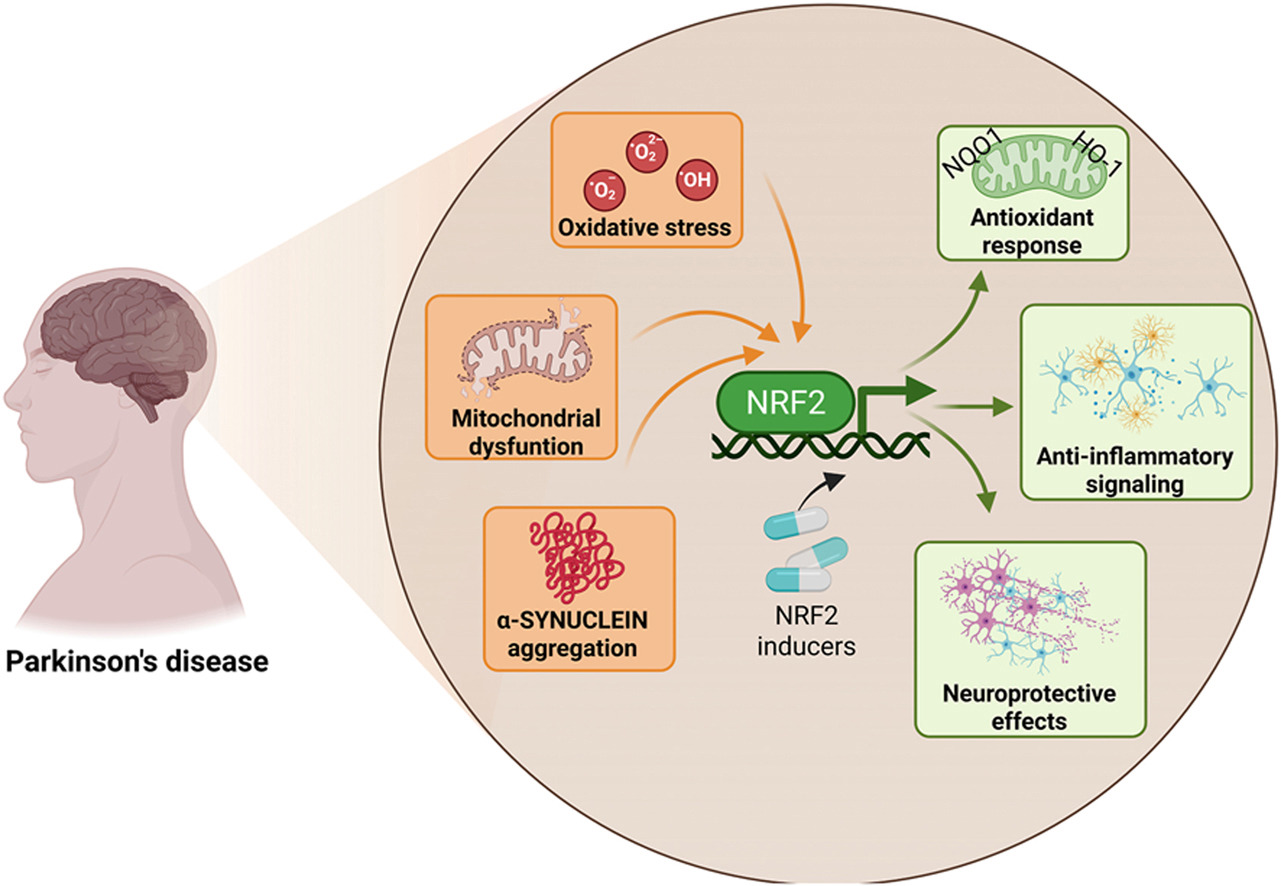
Strategies aimed at restoring or enhancing NRF2 activity hold significant promise as disease-modifying interventions, not only to slow PD progression but also to promote resilience against the broader spectrum of age-associated neurodegenerative and inflammatory conditions.”
https://www.sciencedirect.com/science/article/pii/S0891584926000316 “NRF2 AT THE CROSSROADS OF PARKINSON’S DISEASE AND AGING: MECHANISTIC INSIGHTS AND TRANSLATIONAL PERSPECTIVES”
This review only gave lip service to PD progression outside of the brain, as if the importance of prodromal factors to a person’s neurodegeneration such as dysfunction in gut, eyes, skin, and olfactory systems can be minimized. But failure to recognize early what will doom a person to be unable to recover health in later decades is disingenuous. Since these reviewers omitted early interventions into PD prodromal factors, the best they came up with was interventions to “slow PD progression.”
Maybe these reviewers felt it would be outside the scope of this review to discuss early non-brain PD factors for more than one sentence? However, while PD is defined by striatal brain neurons, Nrf2 activity is much less in brain and central nervous system neurons than elsewhere in the body per Nrf2 Week #2: Neurons.
I disagree with these reviewers’ self-imposed emphasis on aging. Repeating ‘age-associated’ numerous times seemed as if they wanted to influence the reader into thinking age in and of itself was a cause for PD, rather than an imputed mathematical correlation. Their emphasis led to dumb mentions such as senolytics for no apparent reason than senescence is a ‘hallmark of aging’, and to meaningless ‘diseasome of aging’ characterizations, and to ignoring the existence of early non-age-associated PD diagnoses in 20- and 30-year-olds.
Whatever it takes to get published, I’d guess. Or maybe it’s that the number of omissions and useless points a review paper makes increases with the number of reviewers and their sponsors’ agendas.
For example, why was it permissible to dedicate lip service to ‘exposome’ factors like microplastics, environmental pollution, and viruses, but it’s still not permitted in 2026 to discuss research into the impacts on vascular disease and neurodegeneration of lipid nanoparticles and DNA contamination in what a large number of humans were exposed to by injected pharmaceuticals starting in late 2020? Not to mention two studies published in 2024 of over 2.5 million people whose incidences of neurologic issues, mild cognitive impairment, and Alzheimer’s disease rapidly increased after ‘vaccination’?
I’ve mentioned in this blog many times how it’s every human’s choice whether or not we take responsibility for our own one precious life. I suggest, if it’s not too late, do that for your children’s lives, too.