A 2026 review subject was possible involvement of deuterium in TMAO levels, which contrasted with the usual TMAO meme. I’ll curate this paper through an outline of its sections:
“The human gut microbiome plays many essential roles, but an often-overlooked role is to maintain an abundant supply of deuterium depleted (deupleted) nutrients to fuel the host mitochondria. Excess deuterium (heavy hydrogen) damages mitochondrial ATP synthase nanomotors, leading to a decrease in matrix water production with increased reactive oxygen species (ROS) and inefficient ATP production. A microbial metabolite, trimethylamine N-oxide (TMAO) is a powerful signaling molecule whose plasma levels are high in association with many chronic diseases.
In this paper, we present a hypothesis that TMAO is a marker for deuterium overload in the methylation pathway, in addition to its role as an indicator of a disrupted gut microbiome. The original study that brought attention to TMAO involved feeding mice synthetic choline with fully deuterated methyl groups. Fully deuterated TMAO was subsequently detected in the plasma. By contrast, a diet rich in eggs, a natural source of choline (a precursor to TMAO), does not raise TMAO levels. Many of the pathologies that are linked to elevated TMAO can also be viewed as strategies to promote the supply of deupleted water to the mitochondria, systemically.
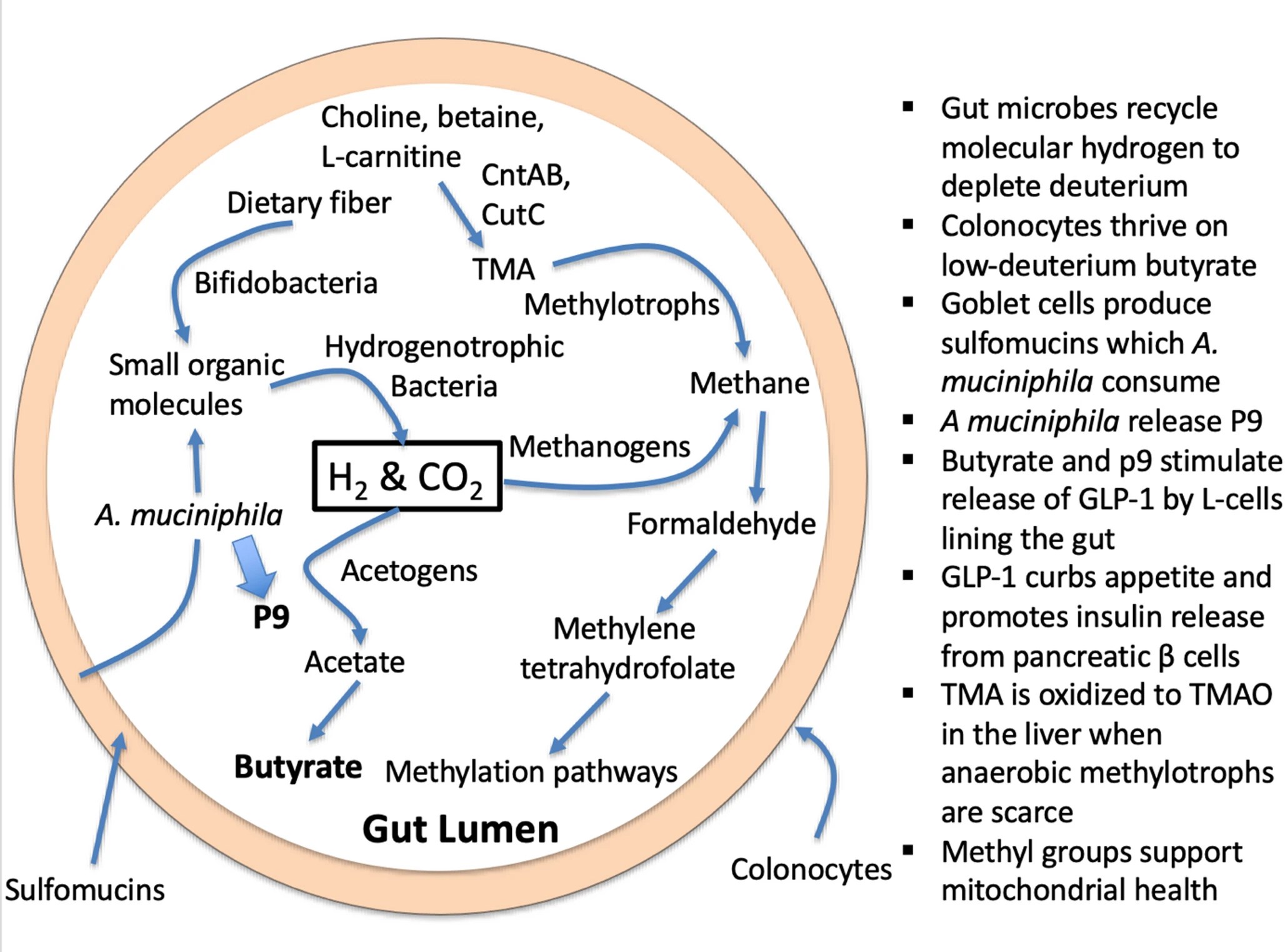
1. Introduction – DNA and histone methylation regulate epigenetic modifications and imprinting. Phosphatidylcholine is a precursor to acetylcholine, an excitatory neurotransmitter in the brain. Trimethylated lysine molecules, recovered during protein metabolism, are precursors to L-carnitine, which facilitates the transport of fatty acids into the mitochondria to be oxidized for fuel production. Dietary phosphatidylcholine and dietary L-carnitine, in addition to endogenous sources, are important nutrients that provide methyl groups to the methylation cycle. Choline and L-carnitine, as well as a close relative, betaine (trimethylglycine), are all precursors to trimethylamine (TMA), a small methylated amine produced through microbial enzymatic action. Obligate anaerobic hydrogen-dependent archaea called methylotrophs in the gut can reduce the three methyl groups in TMA to methane gas.
2. Evidence that deuterium disrupts mitochondrial function – Deuterium (2H) is a heavy isotope of protium (1H; hydrogen), and it is pervasive in nature, found in seawater at a concentration of 155 parts per million (ppm) relative to protium. Deuterium is highly damaging to the F1F0-ATP synthase (ATPase) nanomotors in the mitochondria that produce ATP, the primary fuel source of the cell. Deuterium loading suppresses the activity of many fundamental biologically important hydrolytic enzymes that depend on proton tunneling. It is likely that deuterium increases the frequency of unrepaired nuclear DNA mutations, by suppressing the activity of deuterium-sensitive repair enzymes.
The inherent collective proton tunneling (ICPT) process, which uses membrane-bound ATPase nanomotors in living organisms, is nature’s ultimate tool for discriminating hydrogen isotopes. This is because a deuteron (2H) cannot replace a proton (1H) in its tunnel protein during enzymatic transmembrane transport due to its doubled mass and twice larger atomic nuclear size. The result is large compartmental, inter- and intramolecular deuterium disequilibrium in 2H/1H ratios in all biomolecules, which readily distinguishes respiration from aerobic fermentation with adaptive significance. Deuterons irreversibly clog single proton tunneling ATP synthase nanomotors in mitochondria, resulting in the complete breakdown of ICPT.
This initiates many disease-causing molecular crowding mechanisms, which we review herein from the perspective of prokaryotic proton pumping and H2 gas formation in the organic molecular realm of mitochondrial proton-donating substrates. Understanding at the systems level how humans protect mitochondrial ICPT processes and ATP synthase is a fascinating journey reviewed herein. We hypothesize that the process uses TMAO as the microbial stepping stone, employing deuterium discrimination to become an active player in forming the biological reaction.
2.1 Quantum tunneling and proton-coupled electron transport – ICPT is a theoretical quantum mechanical phenomenon proposing that a single proton spontaneously passes through a potential energy barrier, typically within a hydrogen bond, in a manner that can be functionally irreversible. Unlike classical particles that must surmount energy barriers, protons can ‘tunnel’ through them due to their wave-like nature. Often, this single proton tunneling is part of a larger process where a proton and an electron are transferred simultaneously (or sequentially) as a single kinetic step, often in the presence of strong electric fields that stabilize the transferred state. This process is referred to as ‘proton-coupled electron transport’ (PCET).
Mitochondria exploit PCET to build the proton gradient that powers the nanomotors to produce ATP in the electron transport chain (ETC). Many enzymes, such as dehydrogenases and lipoxygenases, exploit proton tunneling to carry out their reactions. Deuterons are much less capable of tunneling, so this becomes a way to select for substrates containing protons rather than deuterons. The very large kinetic isotope effect (KIE) for soybean lipoxygenase is an example of this phenomenon.
NADH-ubiquinone oxidoreductase (Complex I) couples the transfer of two electrons between NADH and ubiquinone to the translocation of four protons across the membrane. This process provides the driving force for ATP synthase, which harnesses the gradient to produce ATP, but it also assures that few, or no, deuterons arrive on the other (intermembrane) side of the membrane, protecting the ATPase nanomotors.
SAMe, the universal methyl donor, plays a crucial role in regulating oxidative phosphorylation (OXPHOS). It is primarily synthesized in the cytoplasm and imported into the mitochondria via the import protein SAMC. SAMC is the only mitoSAM carrier and is required for OXPHOS and oxidative tricarboxylic acid (TCA) metabolism, showing a strong dependency of mitochondrial health on one-carbon (1C) metabolism. We hypothesize that the importance of SAMe to mitochondrial health is directly linked to the plausible theory that SAMe’s methyl groups are normally highly deupleted.
3. Are microbially synthesized methyl groups and butyrate deuterium depleted? – A careful tracing of multiple metabolic processes taking place in a human cell reveals that they are plausibly designed to greatly restrict the number of deuterons that are in the mitochondrial water. This strategy helps to minimize exposure of the ATPase nanomotors to deuterons. In part, this feat is accomplished through enzymes such as flavoproteins that greatly favor protium over deuterium in their reaction, i.e., that have a high deuterium KIE. The physics usually involves configuring the enzyme to support proton tunneling, since deuterons are much less capable of such tunneling.
Another way to support a reduced deuterium supply to the ATPase nanomotors is to select nutrients that are naturally low in deuterium to feed into the tricarboxylic acid (TCA) cycle. This is what makes the metabolites produced by the gut microbes via hydrogen recycling very significant. The enzyme expressed by anaerobic archaea that metabolize TMA, TMADH, is a flavoprotein with a high deuterium KIE (~ 8.6) due to vibrationally assisted hydrogen tunneling.
This means that the hydrogen recycling that takes place during its metabolism further scrubs deuterium from methylation pathways, while TMA that is left behind becomes enriched in deuterium. This unmetabolized TMA is converted to deuterium-enriched trimethylamine N-oxide (TMAO) in the liver and released into the circulation. Elevated TMAO levels in plasma are associated with increased risk to cardiovascular disease and a long list of other inflammatory diseases, as we will detail in the coming sections.
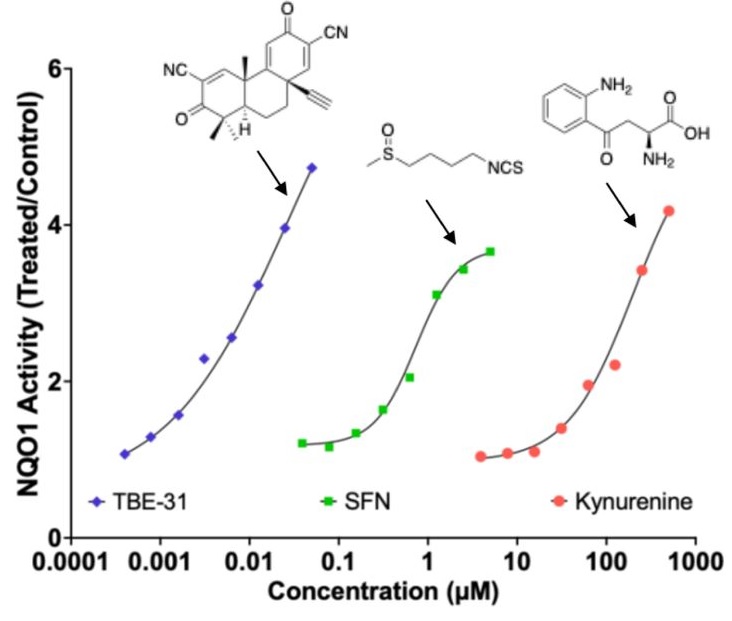
4. Natural and synthetic choline have different effects on TMAO levels: does deuterium play a role? – The original 2011 paper that first identified TMAO as a risk factor for heart disease, involved feeding mice phosphatidylcholine where all the protons in the methyl groups attached to the nitrogen atom were replaced with deuterium, so that the researchers could trace the products of the nutrient in the body. It turns out that they accidentally conducted an experiment testing what happens when phosphatidylcholine is extremely enriched in deuterium. 🙂 They also determined that supplementation of mice with deuterated choline, TMAO, or betaine resulted in upregulation of multiple macrophage scavenger receptors linked to atherosclerosis. TMAO was not produced if the mice were pretreated with antibiotics or in experiments with germ-free mice, confirming that microbial enzymatic action was a necessary precondition.
A paper in 2021 on human subjects compared choline intake from natural dietary sources with supplemental choline bitartrate and found that the latter but not the former raised blood TMAO levels. Notably, these authors wrote in the conclusion of the abstract: ‘Despite high choline content in egg yolks, healthy participants consuming four eggs daily showed no significant increase in TMAO or platelet reactivity.’ However, TMAO levels rose significantly following synthetic choline bitartrate supplementation. This occurred even though the subjects had normal kidney function, showing that elevated TMAO is not just a consequence of kidney disease.
The authors of the original 2011 study had published a follow-on study on human subjects in 2013, in which they supplemented the subjects with D9-PC, essentially repeating the mouse study but with humans as the subjects. They confirmed that D9-TMAO levels were sharply elevated in the plasma and urine following supplementation. Furthermore, an elevated TMAO level predicted an increased risk of major cardiovascular events, after adjustment for traditional risk factors. This study shows that it may not be phosphatidylcholine vs. choline bitartrate that matters, but rather whether the choline is deuterium depleted or deuterium enriched. By contrast, a survey involving over 14,000 participants found that dietary choline protects from both heart disease and stroke. L-carnitine is also a precursor to TMAO, and a mouse study in which the mice were fed deuterated L-carnitine also showed a sharp increase in plasma TMAO following supplementation, further supporting the idea that deuteration is the primary factor promoting TMAO accumulation.
5. TMAO directs metabolism towards deupleting peroxisomal-mitochondrial crosstalk – The synthesis of TMAO from TMA requires hydrogen peroxide. The source of hydrogen peroxide is via peroxisomal fatty acid chain modifications that utilize molecular oxygen dissolved in plasma to produce SCFAs, ketones, NADH and hydrogen peroxide (H2O2). The resulting metabolic water of the reaction is deupleted, as fatty acids are inherently deupleted molecules in biology. Although peroxisomes do not produce ATP directly, they reduce NAD+ for proton delivery to mitochondria via membrane-based intracellular proton transporters. Although the partial contribution of TMAO synthesis to intermediary metabolism is yet to be determined to efficiently deplete deuterium, it is certain that none of the above works well in the deuterium preserving glucogenic metabolic state. There is a strict dependence of peroxisomes on long chain saturated fatty acid substrates with particularly lower deuterium-related chemical mass.
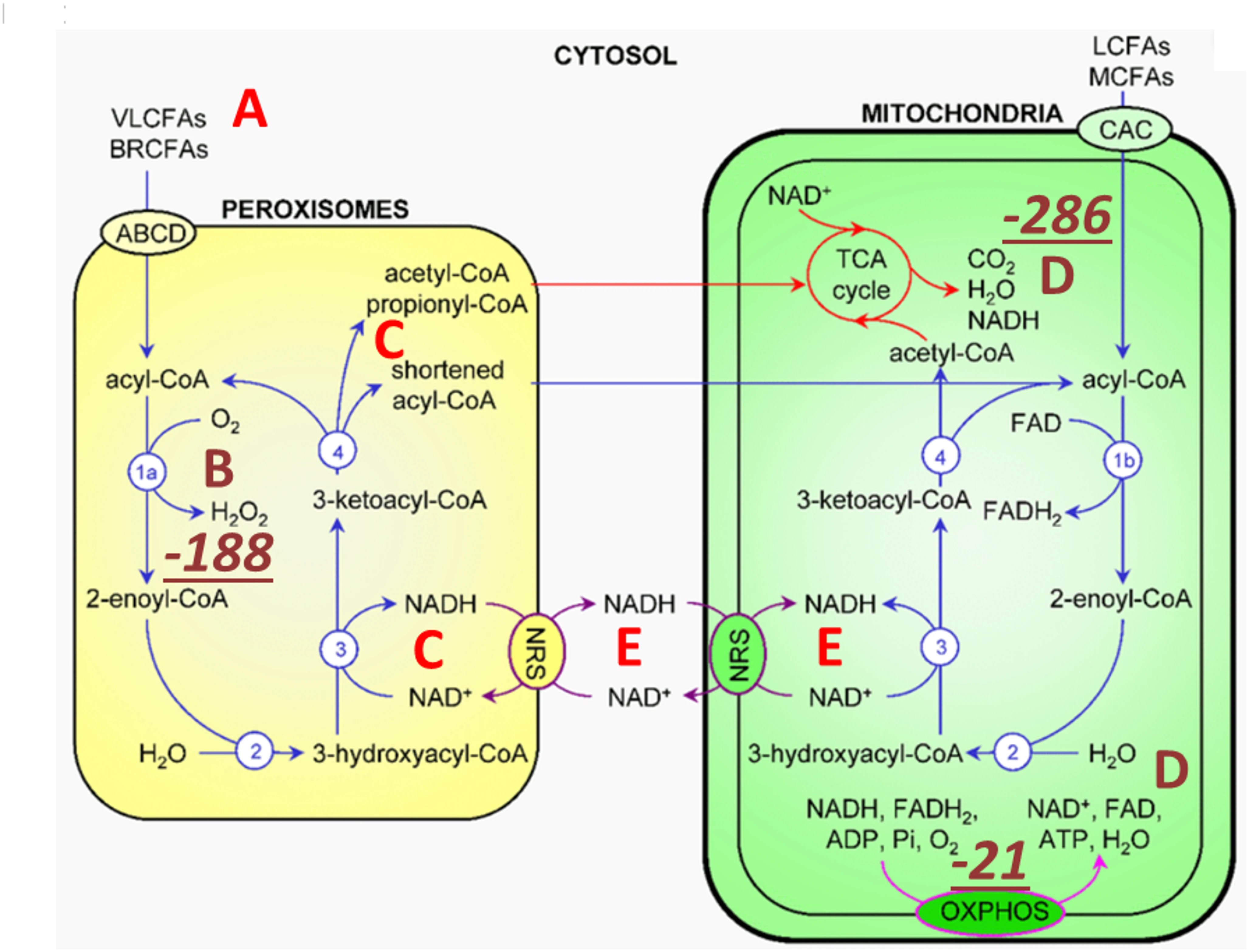
Peroxisomal metabolism triggered by TMAO turnover utilizes very long and branched chain fatty acids (A) as well as dissolved molecular oxygen (B) carried in plasma. Peroxisomes produce SCFAs via β-carbon oxidation, ketones, NADH (C) and hydrogen peroxide (B). H2O2 is rapidly converted to metabolic water by catalase (CAT) that also yields molecular oxygen for the mitochondrial matrix (D) as well as for other cellular compartments. Peroxisomes can also reduce NAD+ for proton delivery to mitochondria via membrane-based intracellular proton transporters. oxidation of very long chain saturated fatty acid β carbons, purportedly of animal source, with the help of molecular oxygen, yields the most deupleted H2O2 by weight. CAT, one of the fastest enzymes in biology with that of isomerases, rapidly and irreversibly produces water from H2O2, while recycling oxygen. Metabolic hydrogen peroxide of fatty acid breakdown with low deuterium consequently provides ATP synthase nanomotor-sparing protons for energy production. High TMAO with hydrogen peroxide turnover can easily depend on CAT-mediated oxygen recycling.
6. Is TMAO an indicator of deuterium overload in the mitochondria?
6.1 TMAO inhibits S-adenosylhomocysteine hydrolase
6.2 TMAO induces reactive oxygen species
6.3 TMAO suppresses autophagy via PI3K/Akt/mTOR activation
7. TMAO and human diseases
8. Does oxidative stress lead to mitochondrial deupletion? – While oxidative stress is a major contributor to cellular damage, the processes involved in resolving ROS are an essential part of the mechanism by which the cell reduces deuterium levels in the mitochondria. Intracellular ROS are derived mainly from NOX, xanthine oxidase, and the mitochondrial electron-transport chain (mETC). Excess mitochondrial deuterium promotes increased ROS generated by the mETC. Superoxide dismutase (SOD) converts ROS to H2O2 , which can release the highly destructive hydroxyl radical in the presence of reduced iron (Fe2+). However, H2O2 is an excellent source of deuterium depleted water (DDW) in the mitochondria, as long as there is sufficient mitochondrial glutathione and both glutathione peroxidase and glutathione reductase are adequately expressed. H2O2 freely crosses the mitochondrial membrane, and, with adequate antioxidant support, it is rapidly converted to two molecules of DDW, catalyzed by glutathione peroxidase.9. Do lipid-laden foam cells support mitochondrial deupletion?
10. Archaeobiotics
11. A crucial role for A. muciniphila
12. Strategies to lower TMAO levels – It is clear that elevated plasma TMAO is a risk factor for a broad range of chronic diseases, and therefore it is compelling that a strategy that reduces plasma TMAO should show health benefits. However, simply avoiding foods that provide precursors to TMAO is not likely to be productive. Choline, L-carnitine, and betaine are the primary sources that fuel the methylation pathway. Eggs and seafood, rich sources of these nutrients, also contain many valuable micronutrients and healthy fats that are also essential.Deuterium depleted water (DDW) is commercially available, at dilution levels as low as 5 ppm. It can be mixed with tap water to simulate natural glacier water, typically containing around 100 ppm deuterium. Although the number of studies on the effects of therapeutic deuterium depletion on various health conditions is small, a review paper found that deuterium depletion has shown promise in preventing and treating cancer, improving long-term memory, enhancing sports performance, and reducing symptoms of depression.
It is apparent that the best way to reduce TMAO levels, while simultaneously boosting methylation supplies, is to promote an abundant colonization of anaerobic archaea in the gut, so that they can clear (fully metabolize) TMA before it has a chance to become TMAO.
13. Discussion – In this paper, we develop the argument that TMAO serves as a marker for excess deuterium in the methylation pathway, and, by extension, in the mitochondria, systemically. While methyl groups have powerful epigenetic effects, the ultimate fate of methyl groups is their metabolism to CO2 and water that is most likely deuterium depleted in the mitochondria. A microbial imbalance leading to reduced colonization by beneficial bacteria and an overgrowth of pathogenic species is the primary cause of overproduction of TMAO.
14. Conclusions – We have shown that TMAO, a causal factor for many diseases, may act as a marker for gut dysbiosis and for excess deuterium load in mitochondria, systemically. We traced through many of the biological pathways involving 1C metabolism and showed the integral role that gut bacteria play in stripping deuterium from methyl groups.”
https://link.springer.com/article/10.1007/s11306-026-02443-3 “The essential role of hydrogen gas recycling by gut microbes in reducing deuterium load in host mitochondria: is trimethylamine oxide a deuterium sensor?”
I take Now brand taurine, acetyl-L-carnitine, flush-free niacin, and betaine. I asked them whether these products are evaluated for their deuterium content. Will update with their response.
Update: I received a response from Now Foods that they don’t analyze their products for deuterium. Fine. I can’t post their response without their permission.
I decided that, of all their products, the one I probably don’t need anymore is betaine (trimethyl glycine). I’ve taken it for about 20 years at below 4 grams daily because at and above that level has lipid-augmenting side effects.
I took it so that my body wouldn’t have to convert choline to betaine during the methionine methylation cycle. Last year I started eating three eggs twice a day, and the year before last, three eggs once daily. I haven’t been concerned about choline for a while.
Determining whether or not I’m making a mistake will have to wait for my next lab test later this year. I’ll reconsider if homocysteine significantly rises or falls. Until then, 82 grams (dry weight) of Avena nuda whole organic oats will be my primary betaine source.