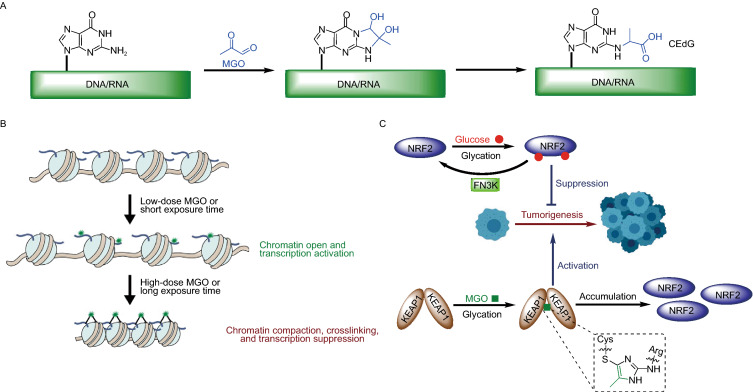
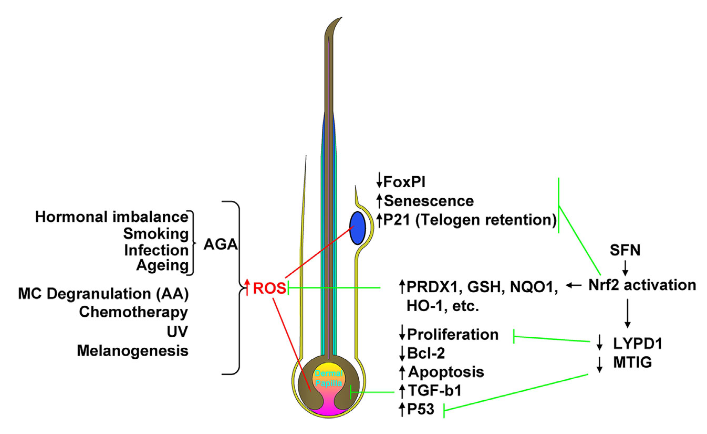
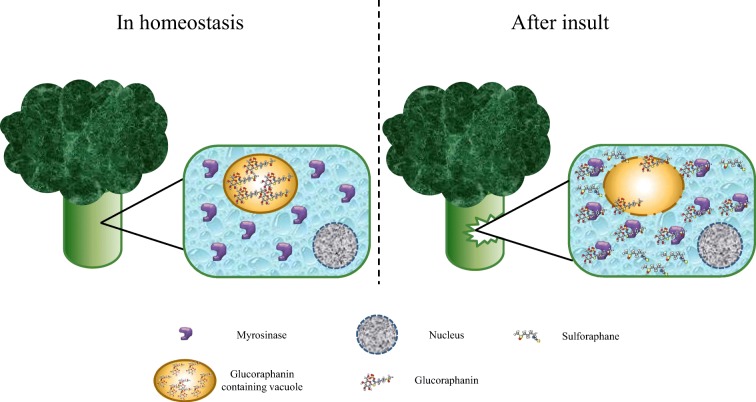
I’m clearing out the below queue of 27 studies and reviews I’ve partially read this year but haven’t taken the time to curate. I have a pesky full-time job that demands my presence elsewhere during the day. :-\
Should I add any of these back in? Let’s be ready for the next decade!
Early life
https://link.springer.com/article/10.1007/s12035-018-1328-x “Early Behavioral Alterations and Increased Expression of Endogenous Retroviruses Are Inherited Across Generations in Mice Prenatally Exposed to Valproic Acid” (not freely available)
https://www.sciencedirect.com/science/article/pii/S0166432818309392 “Consolidation of an aversive taste memory requires two rounds of transcriptional and epigenetic regulation in the insular cortex” (not freely available)
https://www.nature.com/articles/s41380-018-0265-4 “Intergenerational transmission of depression: clinical observations and molecular mechanisms” (not freely available)

https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6454089/ “Epigenomics and Transcriptomics in the Prediction and Diagnosis of Childhood Asthma: Are We There Yet?”
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6628997/ “Placental epigenetic clocks: estimating gestational age using placental DNA methylation levels”
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6770436/ “Mismatched Prenatal and Postnatal Maternal Depressive Symptoms and Child Behaviours: A Sex-Dependent Role for NR3C1 DNA Methylation in the Wirral Child Health and Development Study”
https://www.sciencedirect.com/science/article/pii/S0889159119306440 “Environmental influences on placental programming and offspring outcomes following maternal immune activation”
https://academic.oup.com/mutage/article-abstract/34/4/315/5581970 “5-Hydroxymethylcytosine in cord blood and associations of DNA methylation with sex in newborns” (not freely available)
https://physoc.onlinelibrary.wiley.com/doi/full/10.1113/JP278270 “Paternal diet impairs F1 and F2 offspring vascular function through sperm and seminal plasma specific mechanisms in mice”
https://onlinelibrary.wiley.com/doi/full/10.1111/nmo.13751 “Sex differences in the epigenetic regulation of chronic visceral pain following unpredictable early life stress” (not freely available)
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6811979/ “Genome-wide DNA methylation data from adult brain following prenatal immune activation and dietary intervention”
https://link.springer.com/article/10.1007/s00702-019-02048-2 “miRNAs in depression vulnerability and resilience: novel targets for preventive strategies”
Later life
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6543991/ “Effect of Flywheel Resistance Training on Balance Performance in Older Adults. A Randomized Controlled Trial”
https://www.mdpi.com/2411-5142/4/3/61/htm “Eccentric Overload Flywheel Training in Older Adults”
https://www.nature.com/articles/s41577-019-0151-6 “Epigenetic regulation of the innate immune response to infection” (not freely available)
https://link.springer.com/chapter/10.1007/978-981-13-6123-4_1 “Hair Cell Regeneration” (not freely available)
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6422915/ “Histone Modifications as an Intersection Between Diet and Longevity”
https://www.sciencedirect.com/science/article/abs/pii/S0306453019300733 “Serotonin transporter gene methylation predicts long-term cortisol concentrations in hair” (not freely available)
https://www.sciencedirect.com/science/article/abs/pii/S0047637419300338 “Frailty biomarkers in humans and rodents: Current approaches and future advances” (not freely available)
https://onlinelibrary.wiley.com/doi/full/10.1111/pcn.12901 “Neural mechanisms underlying adaptive and maladaptive consequences of stress: Roles of dopaminergic and inflammatory responses”
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6627480/ “In Search of Panacea—Review of Recent Studies Concerning Nature-Derived Anticancer Agents”
https://www.sciencedirect.com/science/article/abs/pii/S0028390819303363 “Reversal of oxycodone conditioned place preference by oxytocin: Promoting global DNA methylation in the hippocampus” (not freely available)
https://www.futuremedicine.com/doi/10.2217/epi-2019-0102 “Different epigenetic clocks reflect distinct pathophysiological features of multiple sclerosis”
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6834159/ “The Beige Adipocyte as a Therapy for Metabolic Diseases”
https://www.sciencedirect.com/science/article/abs/pii/S8756328219304077 “Bone adaptation: safety factors and load predictability in shaping skeletal form” (not freely available)
https://www.nature.com/articles/s41380-019-0549-3 “Successful treatment of post-traumatic stress disorder reverses DNA methylation marks” (not freely available)
https://www.sciencedirect.com/science/article/abs/pii/S0166223619301821 “Editing the Epigenome to Tackle Brain Disorders” (not freely available)