People will forgive you for being wrong, but they will never forgive you for being right – especially if events prove you right while proving them wrong. Thomas Sowell
A 2026 paper presented results of a clinical trial that selectively added myrosinase enzyme via mustard powder and vitamin C to measure in vivo effects on glucoraphanin conversion to sulforaphane:
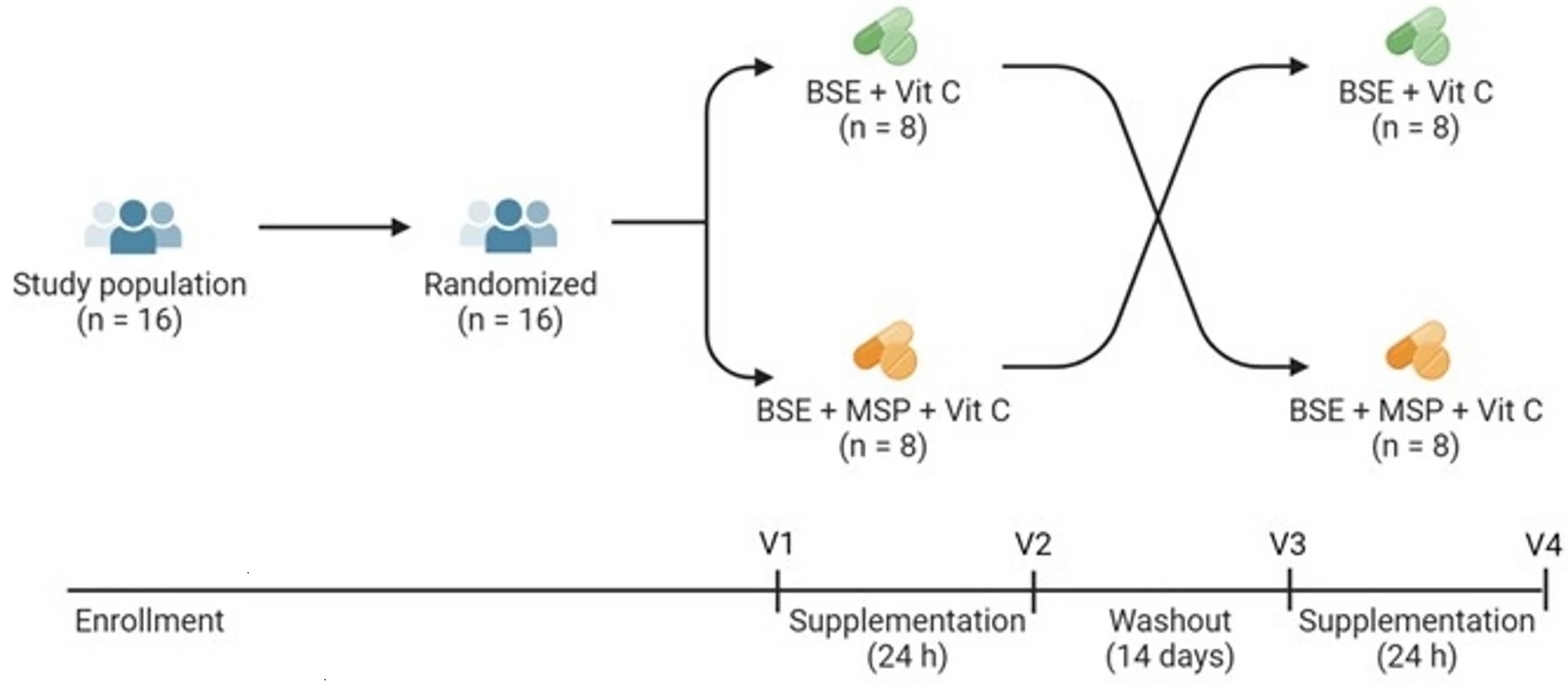
“Effects of exogenous myrosinase (Myr) on conversion efficiency of glucoraphanin (GR) to sulforaphane (SF) was compared to gut microbial Myr-like activity. In a randomized, double-blind, crossover study, sixteen subjects (9 F: 7 M) received a single oral dose of GR in 385 mg broccoli seed extract (BSE) with 72.5 mg Myr-containing mustard seed powder, or broccoli seed extract alone, both with 100 mg ascorbic acid.
GR + Myr, on average, doubled the bioavailability of SF (39.8 ± 3.1%) compared to GR alone (18.6 ± 3.1%), and increased the conversion rate in the first 8 h (25.4% ± 2.7%) compared to GR alone (8.0% ± 2.7) based on measurement of urinary metabolites. The majority of subjects given GR as BSE with exogenous Myr and Vit C, converted GR to SF notably faster (e.g., within the first 8 h), than those given GR and Vit C alone.
One of the most likely explanations for the pronounced differences is that when SF was produced as a result of added exogenous Myr, it was produced and absorbed in the small intestine and metabolized primarily to its glutathione (GSH) derivatives and excreted in urine. This is a more rapid process than when GR passes into the large intestine and then is acted upon by the greater bacterial population within that terminal segment of the gastrointestinal system.
No differences were observed in the 8 to 24 h urine collection (Time 24 h) between the two treatments: 11.7 ± 1.3% for GR + Myr vs. 8.9 ± 1.3% for GR alone. Bacterial communities did not differ between low/high GR converters following supplementation.
Many bacteria which persist in the small intestine and upper large intestine, as well as in mucosal-associated fractions, are not well represented in feces. The lack of anatomically specific microbial communities in the human gut limits our knowledge of GR conversion in people.”
https://www.nature.com/articles/s41598-026-39389-4 “Exogenous myrosinase from mustard seed increases bioavailability of sulforaphane from a glucoraphanin-rich broccoli seed extract in a randomized clinical study”
1. Lost in this study’s shuffle was the reason why sulforaphane’s effects are beneficial in the first place. As Switch on your Nrf2 signaling pathway pointed out:
“We use N-acetylcysteine (NAC) in the lab all the time because it stops an Nrf2 activation. So that weak pro-oxidant signal that activates Nrf2, you switch it off by giving a dose of NAC. It’s a potent antioxidant in that right, but it’s blocking signalling. And that’s what I don’t like about its broad use.”
It’s relevant to the Nrf2 activating effects of sulforaphane when antioxidant vitamin C taken to increase myrosinase hydrolyzation of glucoraphanin to sulforaphane if this vitamin C dose may also block Nrf2 activation. An increase of sulforaphane and its metabolites wouldn’t physiologically matter if their beneficial effects were simultaneously blocked.
2. Another indicator that these researchers lost the plot was shown when they asserted: “These data suggest that the present study may not have included sufficient AA to optimize Myr enzyme activity in vivo, another consideration for future studies” based on comparing the in vitro Reference 21 ascorbic acid doses (0, 11, 44, 88, or 154 mg AA/capsule). I don’t have access to Reference 21 to see whether it also didn’t assess Nrf2 activation.
3. For comparison of this study’s 50 mg glucoraphanin dose on two non-consecutive days, the cited Our model clinical trial for Changing to a youthful phenotype with broccoli sprouts provided daily 30 grams of fresh broccoli sprouts that contained an estimated 51 mg of glucoraphanin for ten weeks. Although no broccoli sprout preparation or intake guidelines were enforced, unassisted glucoraphanin conversion to sulforaphane had many beneficial effects in that trial.
4. These researchers stated “No differences were observed in the 8 to 24 h urine collection (Time 24 h) between the two treatments.” Whether the faster small intestine absorption of sulforaphane had benefits over the slower large intestine absorption wasn’t investigated.
5. I’ve previously corresponded with one of this study’s coauthors, and think their research group could do better work without the retired broccoli sprout expert. Maybe they would take more confident ownership of their work if they let him ride off into the sunset, and decide for themselves what predictable findings would be in their research effort’s scope?
Maybe they wouldn’t let fester the same old issues in his papers? It doesn’t say anything good about current research if its main findings just repeat last decade’s findings.
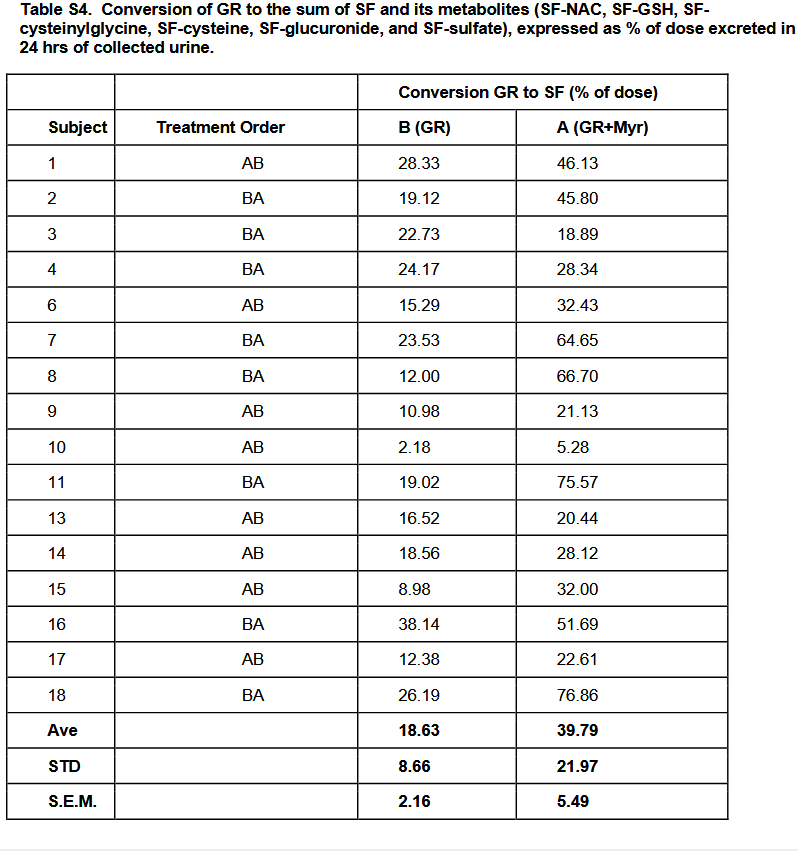
For example, Table S4 has stories that weren’t told. It conforms to the expert’s cited 2015 study findings, but presenting averages doesn’t reveal causes for individual differences. So questions on the individual level continue to be unanswered, such as:
Subject 8 had more than a fivefold increase of glucoraphanin-only conversion to glucoraphanin + myrosinase conversion. What blocked the other subjects from achieving similar results?
Why was Subject 10 so far behind everyone else’s capabilities? They would have had to increase their glucoraphanin-only conversion by fourfold just to get to the next lowest person’s level, Subject 15, but sixfold to get to Subject 15’s glucoraphanin + myrosinase conversion level.
6. Every researcher wants to have an impact on their field. The time to think over how to newly research possible impactful outcomes is before the study starts.
Since I’m in my seventh year of eating broccoli sprouts every day, I would have paid close attention to more rigorous bioavailability, more exact microbiota collection techniques, or detailed exploration of differences in people’s responses to the same treatments, all of which were predictable issues beforehand. I didn’t really care for listing them in the Discussion section, then dismissing investigations of these findings for some future research to explore.
1. I’ve continued daily practices from Year Five to experience another year without being sick (if I don’t count getting MSG poisoning from Chinese food.) I consequently scheduled a doctor visit next week to get a sumatriptan prescription refilled.
– In that post’s comments, Ole Bisgaard Pedersen asked if I took NAD+ supplements such as NAM, NMN or NR – forms of vitamin B3 that are precursors to NAD+. I didn’t note that last year I started taking Now brand Flush-free niacin 500 mg mid-morning.
Nicotinamide riboside has the most human evidence, including a Tru Niagen clinical trial that showed improvements in peripheral artery disease. It’s too expensive for long-term use, though.
Even if I could afford it, there isn’t a magic bullet for fixing vascular system dysfunction. PAD is just one symptom of a cardiovascular system that needs to be overhauled then maintained at a healthy level. There is no clinical trial that has a logical therapeutic end point to stop treatments where a person could say, “I’ve done enough for my vascular system, my physical and cognitive functions won’t backslide.”
– 2-3 years ago, I changed from microwaving broccoli sprouts in a plastic bag to microwaving them in a small bowl with a small plate covering it to keep them from popcorning out of the bowl. I use a 1000W microwave oven on 80% power for ten seconds.
3. The two vitamin C macaque studies I’ve recently curated both ran for a human equivalent of ten years. I was encouraged that both found Nrf2 activation to be part of their causal beneficial evidence, since vitamin C wasn’t on my radar as a Nrf2 activator.
I expect that in four years I’ll write a Year Ten post on eating microwaved broccoli sprouts. I haven’t seen human evidence for broccoli extracts that bypass small intestine absorption and metabolism per Glucosinolate and isothiocyanate human interventions, or enteric capsules, or nanoformulations as suitable substitutes. Maybe studies on broccoli sprout powder or a Nrf2 activator that tops sulforaphane will be published before then, who knows.
A 2025 human study of four geographically distinct populations investigated inflammation biomarkers:
“Inflammaging, an age-associated increase in chronic inflammation, is considered a hallmark of aging. However, there is no consensus approach to measuring inflammaging based on circulating cytokines.
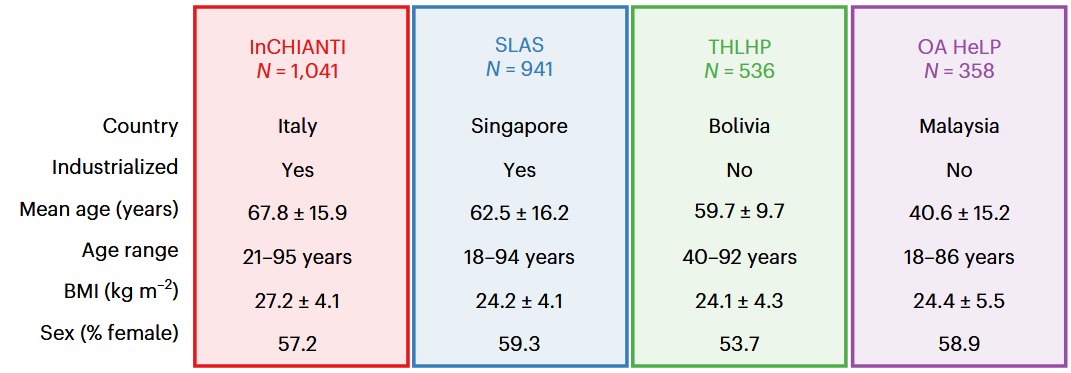
We assessed whether an inflammaging axis detected in the Italian InCHIANTI dataset comprising 19 cytokines could be generalized to a different industrialized population (Singapore Longitudinal Aging Study) or to two indigenous, nonindustrialized populations: the Tsimane from the Bolivian Amazon and the Orang Asli from Peninsular Malaysia.
Much cytokine variation in these populations is probably due to the type and severity of current infections, not aging. Our results show that cytokines are not destiny with regard to inflammaging and chronic disease.
Even within industrialized populations, manifestation of inflammaging is highly heterogeneous. It may reflect immune dysregulation resulting from an evolutionary mismatch of physiology and environment, aligning with the notion that the hallmarks of aging are not universals, but rather common manifestations whose importance varies by context.
The Singapore Longitudinal Aging Study was similar to InCHIANTI except for IL-6 and IL-1RA. The Tsimane and Orang Asli showed markedly different axis structures with little to no association with age and no association with age-related diseases.
Inflammaging, as measured in this manner in these cohorts, appears to be largely a byproduct of industrialized lifestyles, with major variation across environments and populations.”
This study used the terms “industrialized” and “nonindustrialized” two dozen times without defining either, as if every reader knew what these researchers meant. Impreciseness wasn’t an accident, though. It invokes a meme rather than promote reader understanding. They did the same thing with not defining a significant biomarker, IL-IRA.
I’ll highlight one of the unmeasured and potentially important differences among these four populations, daily sunlight exposures. Although Singapore is one degree of latitude above the equator, sunlight availability doesn’t matter when the average 62-year-old retiree spends their days mostly indoors, and their nights viewing blue-light screens. What would be “industrialized” about this lifestyle?
Here are five 2025 human ergothioneine studies, starting with a clinical trial of healthy older adults:
“In this 16-week randomized, double-blind, placebo-controlled trial, 147 adults aged 55–79 with subjective memory complaints received ergothioneine (10 mg or 25 mg/day ErgoActive®) or placebo. Across all the groups, approximately 73% of participants in each group were female, with a median age of 69 years.
The primary outcome was the change in composite memory. Secondary outcomes included other cognitive domains, subjective memory and sleep quality, and blood biomarkers. At baseline, participants showed slightly above-average cognitive function (neurocognitive index median = 105), with plasma ergothioneine levels of median = 1154 nM.
Although not synthesized in the human body, ergothioneine is efficiently absorbed via the OCTN1 transporter (also known as the ergothioneine transporter, or ETT), which is expressed in many tissues, including the intestine, red blood cells, kidneys, bone marrow, immune cells, skin, and brain. This transporter enables ergothioneine to accumulate in high concentrations in organs vulnerable to oxidative stress and inflammation. Ergothioneine has multiple cellular protective functions, including scavenging reactive oxygen species, chelating redox-active metals, suppressing pro-inflammatory signaling, and protecting mitochondrial function.
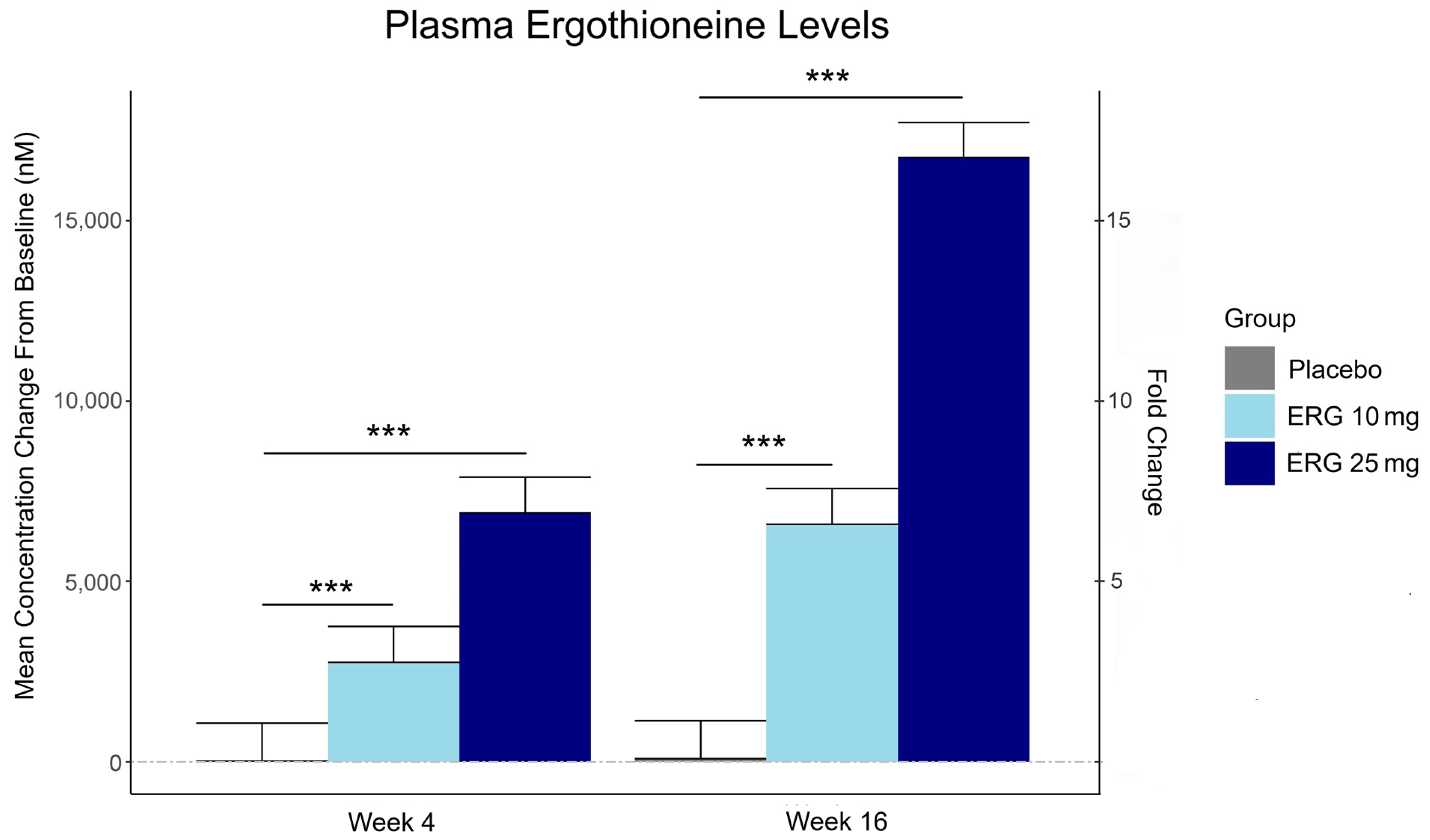
Plasma ergothioneine increased by ~3- and ~6-fold for 10 mg, and ~6- and ~16-fold for 25 mg, at weeks 4 and 16, respectively.
While the primary outcome, composite memory, showed early improvement in the 25 mg group compared to baseline, this effect was not sustained and did not differ from placebo. Reaction time showed a significant treatment-by-time interaction favoring ergothioneine, yet the between-group differences were not significant, suggesting that any potential benefits were modest and require validation in larger or longer studies.
Other cognitive effects observed were primarily within-group and not consistently dose-responsive, highlighting the challenge of detecting objective cognitive changes over a relatively short study duration in high-functioning healthy populations. However, positive effects of ergothioneine supplementation were observed on subjective measures of prospective memory and sleep initiation that were not seen in the placebo group.
This trial adds to the growing body of evidence supporting the favorable safety profile of ergothioneine. No adverse events attributable to ergothioneine were reported. Additionally, we observed potential hepatoprotective effects, with significant reductions in the plasma AST and ALT levels, particularly among males in the ERG 25 mg group.”
https://www.mdpi.com/1661-3821/5/3/15 “The Effect of Ergothioneine Supplementation on Cognitive Function, Memory, and Sleep in Older Adults with Subjective Memory Complaints: A Randomized Placebo-Controlled Trial”
The third graphic for Ergothioneine dosing, Part 2 showed a human study where a 25 mg dosing stopped after Day 7, but the plasma ergothioneine level stayed significantly higher than baseline through Day 35.
The second graphic for Ergothioneine dosing, Part 2 was a male mouse experiment where plasma ergothioneine levels of a human equivalent 22 mg to 28 mg daily dose kept rising through 92 weeks.
This trial couldn’t explain the desirability of a 25 mg daily dose that was likely (per the second and third graphics for Ergothioneine dosing, Part 2) to sustain the subjects’ increased plasma ergothioneine levels well after the trial ended at Week 16. What effects can be expected from a sustained plasma ergothioneine level that’s 16 times higher than the subjects’ initial levels? Were these 16-fold sustained plasma ergothioneine levels better or worse than the 6-fold increases in the 10 mg group, both of which were likely to continue past the trial’s end?
A representative of the trial’s sponsoring company talked a little more about the trial in this interview:
Another clinical trial investigated ergothioneine’s effects on skin:
“We conducted an 8-week, randomized, double-blind, placebo-controlled clinical trial to evaluate effects of daily oral supplementation with 30 mg of ergothioneine (DR.ERGO®) on skin parameters in healthy adult women aged 35–59 years who reported subjective signs of skin aging. Objective measurements including melanin and erythema indices, skin glossiness, elasticity, and wrinkle and pigmentation counts were used to comprehensively evaluate changes in skin condition.
The OCTN1 transporter is preferentially expressed in basal and granular epidermal layers, where cellular renewal and barrier maintenance are most active. Once internalized, ergothioneine localizes to mitochondria, where it directly scavenges reactive oxygen species (ROS) and protects mitochondrial DNA from UV- and inflammation-induced damage.
At the signaling level, ergothioneine activates key protective pathways such as the Nrf2/ARE axis, enhancing expression of antioxidant enzymes including HO-1, NQO1, and γ-GCLC. These enzymes contribute to redox homeostasis and glutathione regeneration, reinforcing cellular defense systems against photoaging and environmental insult.
In parallel, ergothioneine modulates the PI3K/Akt/Nrf2 and SIRT1/Nrf2 pathways, which are implicated in collagen preservation, inflammation resolution, and mitochondrial maintenance. These pathways converge to reduce matrix metalloproteinase (MMP) activity, enhance collagen synthesis, and suppress pro-inflammatory cytokines (TNF-α, IL-6, IL-1β), all of which are central to maintaining skin structure and function.
Compared to placebo, the DR.ERGO® ergothioneine group showed significantly greater improvements in melanin and erythema reduction, skin glossiness, elasticity, and wrinkle and spot reduction. No adverse events were reported.
These findings corroborate and extend previous clinical evidence from (Hanayama et al., 2024), who investigated an ergothioneine-rich mushroom extract (Pleurotus sp., 25 mg ergothioneine/day) in a 12-week randomized double-blind trial, and (Chunyue Zhang, 2023), who examined pure ergothioneine supplementation (25 mg/day) in a 4-week open-label study. We contextualized our results within this existing literature by comparing key outcomes.
Several limitations should be acknowledged:
The study cohort consisted solely of Japanese women aged 35–59 years, which may limit generalizability across sexes, ethnicities, and age groups.
The 8-week intervention period, while sufficient to detect short-term effects, does not allow conclusions about the sustainability of benefits or the risk of relapse upon discontinuation.
The placebo group also showed modest improvements in self-perception, highlighting the well-documented placebo response in beauty and wellness studies.
This study focused on a single daily dosage (30 mg/day) without evaluating dose–response relationships, and hydration-specific endpoints such as corneometry or transepidermal water loss (TEWL) were not included.”
Two clinical trials investigated ergothioneine’s effects on sleep quality:
“A four-week administration of 20 mg/day ergothioneine (EGT), a strong antioxidant, improves sleep quality; however, its effect at lower doses remains unclear. This study estimated the lower effective doses of EGT using a physiologically based pharmacokinetic (PBPK) model in two clinical trials.
In Study 1, participants received 5 or 10 mg/day of EGT for 8 weeks, and their plasma and blood EGT concentrations were measured. An optimized PBPK model incorporating absorption, distribution, and excretion was assembled. Our results showed that 8 mg/day of EGT for 16 weeks was optimal for attaining an effective plasma EGT concentration.
In Study 2, a randomized, double-blind, placebo-controlled study, participants received 8 mg/day EGT or a placebo for 16 weeks. The subjective sleep quality was significantly improved in the EGT group than in the placebo group.
In mammals, EGT is not generated in the body but is acquired from the diet via the carnitine/organic cation transporter OCTN1/SLC22A4. Its plasma concentration after oral administration is quite stable and gradually increases after repeated dosing on a multi-day basis.
Blood concentrations of EGT increase after Day 8 when EGT intake is interrupted, and they continue to increase until Day 35. The delayed increase in EGT concentration in the blood, compared with that in the plasma, can be interpreted as its efficient uptake by undifferentiated blood cells, which express high levels of OCTN1/SLC22A4 in the bone marrow, and subsequent differentiation to mature blood cells that enter the circulation. This may imply the nonlinear absorption, distribution, and excretion of EGT owing to saturation of the transporter at higher concentrations, potentially leading to difficulty in model construction.
This is the first study to propose a strategy to estimate lower effective doses based on the PBPK model.”
The bolded section above referenced a 2016 study / third graphic for Ergothioneine dosing, Part 2, where a 25 mg dosing stopped after Day 7, but the plasma ergothioneine level stayed high through Day 35. I didn’t see that the referenced 2004 and 2010 studies addressed this 2016 finding.
I also didn’t see that this study’s mathematical model accounted for saturation of the OCTN1 transporter or other effects, such as a very small ergothioneine clearance rate. Okay, lower the ergothioneine dose, and achieve a lower persistent plasma ergothioneine level, to what benefit?
“The present study demonstrated that OCTN1 is associated with myeloid cells rather than lymphoid cells, and especially with erythroid-lineage cells at the transition stage from immature erythroid cells to peripheral mature erythrocytes.”
Persistent high ergothioneine levels aren’t costless. Skewing bone marrow stem cells and progenitor cells toward a myeloid lineage is done at the expense of lymphocytes, T cells, B cells, and other lymphoid lineages.
Where are the studies that examine these tradeoffs? Subjective sleep quality in this study and sleep initiation in the first study above aren’t sufficiently explanatory.
A study investigated associations of plasma ergothioneine levels and cognitive changes in older adults over a two-year period:
“Observational studies have found that lower plasma levels of ergothioneine (ET) are significantly associated with higher risks of neurodegenerative diseases. However, several knowledge gaps remain:
Most of the above studies were based on cross-sectional study design, and potential reverse causation cannot be excluded. It has been suggested that plasma ET declines concomitantly with the deterioration of cognitive function.
Since the impact of a single dietary factor on health is mild, it is prone to be affected by the baseline characteristics of subjects (such as sex, educational level, disease status and gene polymorphism). However, no study has systematically evaluated potential effect modifiers on the association between ET levels and cognitive function.
The dose-response distribution between ET and cognitive function remains undetermined.
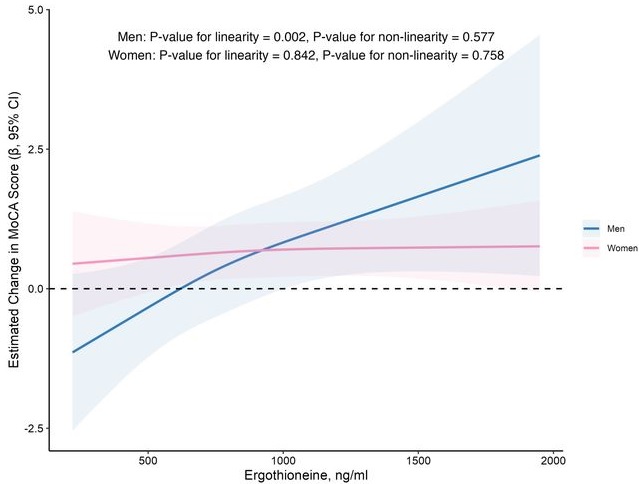
In this prospective cohort study of 1,131 community-dwelling older adults (mean age 69 years), higher baseline plasma ET levels were significantly associated with slower cognitive decline, as assessed by Montreal Cognitive Assessment (MoCA) scores, during a 2-year follow-up period.
When the plasma concentration of ET exceeds 1,000 ng/mL, the decline in cognitive function significantly slows down. However, this association has only been observed in men.
Domain-specific analysis found that the observed ET-MoCA association was mainly driven by the temporary slowdown in the decline of visuospatial/executive and delayed recall. Impaired delayed recall represents one of the earliest and most sensitive cognitive markers of dementia progression, predictive of conversion from MCI to dementia. The preferential preservation of this function by ET suggests targeted neuroprotective effects within the hippocampus.
Visual inspection of the spline curves revealed a potential plateauing effect at ET concentrations ≥1,000 ng/mL in the total population.
Baseline ET concentrations differed between men and women. Most men (81.5%) had concentrations below 1,000 ng/mL (median 754.2, IQR 592.0–937.9 ng/mL). Women exhibited substantially higher median plasma ET concentrations than men, with 35.7% of women exceeded 1,000 ng/mL (median 890.1, IQR 709.7–1,095.6 ng/mL).
Our study included only participants with normal cognitive function, and the results remained robust even after excluding those with baseline cognitive function at the lower end of the normal range. Collectively, our findings support that low ET intake occurs prior to cognitive decline.
Our findings indicate that higher plasma ET levels are significantly associated with slower cognitive decline independent of confounders in non-demented community-dwelling elderly participants, with such association observed in men but not women. Dose-response curves indicated plateauing effects above 1000 ng/mL.”
The average age of this study and the first trial above were both 69 years. Since the first trial’s participants showed slightly above-average cognitive function (neurocognitive index median = 105), with plasma ergothioneine levels of median = 1154 nM at baseline, and this study showed plateauing effects above 1000 ng/mL, I wonder how raising plasma ergothioneine levels above 1000 ng/mL could possibly show a net benefit for older people? What are the trade-offs for older people between potentially increasing slightly above-average cognitive function with ergothioneine and its other effects from saturating their OCTN1 transporter?
This study is at its preprint stage. I’m interested to see if its peer review prompts these researchers to also investigate the common finding that people who are most deficient at baseline have the greatest improvements. If so, would these sex-specific associations still hold?
Wrapping up with a study that investigated associations of serum ergothioneine levels with the risk of developing dementia:
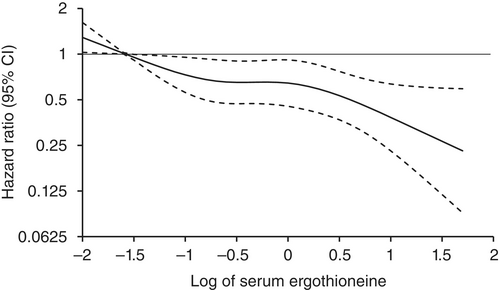
“1344 Japanese community-residents aged 65 years and over, comprising 765 women and 579 men, without dementia at baseline were followed prospectively for a median of 11.2 years.
During follow-up, 273 participants developed all-cause dementia. Among them, 201 had Alzheimer’s disease (AD) and 72 had non-Alzheimer’s disease (non-AD) dementia.
Age- and sex-adjusted hazard ratios (HRs) for all-cause dementia, AD, and non-AD dementia decreased progressively across increasing quartiles of serum ergothioneine. These associations remained significant after adjustment for a wide range of cardiovascular, lifestyle, and dietary factors, including daily vegetable intake.
In subgroup analysis, association between serum ergothioneine levels and the risk of dementia tended to be weaker in older participants and in women:
In older individuals, cumulative burden of multiple risk factors such as hypertension, diabetes mellitus, and smoking may contribute to both neurodegenerative and vascular pathology, potentially diminishing the relative influence of ergothioneine.
In women, postmenopausal hormonal changes, particularly the decline in estrogen, have been associated with increased oxidative stress and a higher vulnerability to neurodegenerative changes.
Several limitations should be noted:
Since serum ergothioneine levels and other risk factors were measured only at baseline, we could not evaluate the changes of serum ergothioneine levels during the follow-up period. Lifestyle modifications during follow-up could have influenced serum ergothioneine levels and other risk factors. In addition, serum ergothioneine level was measured only once, and from a sample.
We cannot rule out residual confounding factors, such as other nutrients in mushrooms and socioeconomic status.
There is a possibility that dementia cases at the prodromal stage were included among participants with low serum ergothioneine levels at baseline.
We are unable to specify which mushroom varieties were predominantly consumed by participants in the town of Hisayama.
Given the limited discriminative ability of serum ergothioneine and potential degradation due to long-term sample storage, we were unable to explore a clinically meaningful threshold value of serum ergothioneine.
Generalizability of findings was limited because participants of this study were recruited from one town in Japan.
These findings suggest that the potential benefit of ergothioneine may be attenuated in individuals with pre-existing, multifactorial risk profiles for dementia.
Our findings showed that higher serum ergothioneine levels were associated with a lower risk of developing all-cause dementia, AD, and non-AD dementia in an older Japanese population. Since ergothioneine cannot be synthesized in the human body, a diet rich in ergothioneine may be beneficial in reducing the risk of dementia.”
For five years I got most of my estimated 7 mg daily ergothioneine intake from mushrooms in AGE-less chicken vegetable soup per Ergothioneine dosing. The soup was always boring, but I got too bored this year and stopped making it. I haven’t replaced mushroom intake with supplements.
I still don’t eat fried or baked foods, preferring sous vide and braising cooking methods to avoid exogenous advanced glycation end products. I avoid buying foods that evoke a hyperglycemic response or otherwise form excessive endogenous AGEs per All about AGEs.
Continuing Part 1 with three 2025 papers, starting with a rodent study of dietary mussel plasmalogens’ effects on atherosclerosis:
“The purpose of this study was to clarify the underlying mechanisms of Mytilus edulis-derived plasmalogens (Pls) against atherosclerosis (AS) in ApoE−/− mice induced by a high-fat diet (HFD), through a comprehensive analysis of hepatic metabolomics and aortic transcriptomics data. Besides Pls role as the storage pool of n-3 PUFAs, the structural feature of vinyl ether bond at sn-1 position confers multiple advantages upon Pls compared to their diacyl counterparts, including enhanced antioxidant capacity, increased membrane fluidity, as well as improved stability and stability of biomembranes.
The C57BL/6 mouse strain is susceptible to high-fat diet (HFD)-induced AS lesions, and ApoE knockout accelerates AS development. Molecular mechanisms by which Pls ameliorate AS were investigated through a comprehensive analysis of hepatic metabolomics and aortic transcriptome profiles, focusing on changes in gene related to the p38 mitogen-activated protein kinase (MAPK) signaling pathway and the downstream inflammatory response.
The concentration of Pls in mussel tissues is 32 μgmg−1 (dry weight), and the obtained Pls contains 49.53% of phosphatidylethanolamine-Pls, 35.87% of phosphatidylcholine-Pls, and 14.60% of phosphatidylserine-Pls. The main fatty acid compositions of Pls are presented in Supplementary Table 1, which indicates that EPA accounts for 45.82% and the n-3/n-6 ratio is 3.84.
Pls inhibited aortic lipid accumulation, prevented thickening of the aortic wall, and suppressed collagen accumulation at the aortic-heart junction. Pls inhibited HFD-induced loosening of hepatocyte arrangement, vacuolization, and accumulation of lipid droplets.
Although several key components of MAPK signaling pathway were suppressed at both the transcriptional and protein levels in Pls-treated mice, no significant changes in phosphorylated p38 protein were observed among the experimental groups in our study. Further research is needed to elucidate the overall inhibitory mechanism of Pls on p38 protein and the MAPK signaling pathway.”
A rodent / human cell study investigated effects of plasmalogens in innate immune system macrophages on atherosclerosis:
“We demonstrate that simultaneous inactivation of two key enzymes involved in macrophage polyunsaturated fatty acid (PUFA) metabolism—ELOVL5, which elongates long-chain PUFAs, and LPCAT3, which incorporates them into phospholipids—disrupts membrane organization by promoting the formation of cholesterol-enriched domains. This increases macrophage sensitivity to cytotoxic oxysterols and leads to more vulnerable atherosclerotic plaques with enlarged necrotic cores in a mouse model of atherosclerosis.
We identified ELOVL5 as one elongase facilitating the conversion of C20 to C22 PUFA. In humans, analysis of 187 carotid plaques reveals a positive correlation between LPCAT3/ELOVL5-generated phospholipids—including arachidonate (C20:4 n-6)-containing ether lipids—and more stable plaque profiles. Additionally, Mendelian randomization analysis supports a causal relationship between LPCAT3 expression and reduced risk of ischemic stroke.
Potentially beneficial effects we observed in mice and in human atheroma plaques were mainly associated with PLs enriched in omega-6, particularly in AA. Although omega-6 FAs are often considered as pro-inflammatory, their role is undergoing reconsideration, with markers linked to the intake of omega-6 appearing beneficial in the context of cardiovascular diseases. In this context, it is worth to note that AA-containing plasmalogens have been previously identified as markers of healthy obesity.
Our findings uncover a regulatory circuit essential for PUFA-containing phospholipid generation in macrophages, positioning PUFA-containing ether lipids as promising biomarkers and therapeutic targets.”
A human study included plasmalogens in investigating associations among people with mental illness and their lipid profiles:
“Plasma lipidomic profiles of 623 individuals (188 schizophrenia (SCZ), 243 bipolar disorder (BD), 192 healthy controls) belonging to the PsyCourse Study were assessed using liquid chromatography and untargeted mass spectrometry. Exact etiology of these major mental health disorders is yet unknown and while their symptoms overlap, their diagnostic criteria are based on clinical evaluations of symptoms without objective markers.
Cognitive dysfunction is among the most disabling symptoms of SCZ and BD, and is difficult to treat with the commonly used pharmacologic regimes. Consequently, it has important impacts on long-term functional outcomes.
We aimed to answer the question, whether specific lipid species or classes were associated with differential performance across various cognitive domains, including psychomotor and processing speed, executive function, short-term and working memory and crystalized intelligence and whether these associations were affected by diagnoses.
Lipids belonging to the phosphatidylethanolamine plasmalogen (PE-P) class emerged as the main lipid class associated negatively with DG-SYM test performance, representative of processing and psychomotor speed. Our findings showed that higher levels of PE-P 42:5, PE-P 40:4, PE-P 40:5, and ceramide 38:1 in plasma samples of our study are significantly associated with poorer DG-SYM test performance. The DG-SYM test mainly measures processing speed, the amount of time required to complete a series of cognitive tasks. Enrichment analysis also showed significant associations between other lipid classes and various cognitive tests.
Our findings suggest a link between lipids and cognitive performance independent of mental health disorders. Independent replication is warranted to better understand if phosphatidylethanolamines could represent an actionable pharmacologic target to tackle cognitive dysfunction, an important unmet clinical need that affects long-term functional outcomes in individuals with severe mental health disorders.”
It was apparently beyond these researchers’ expertise to offer informed discussion on this study’s associative link between enrichment of these three phosphatidyl ethanolamine plasmalogens and cognitive dysfunction. Grok countered that their depletion was associated with neurodegenerative diseases (Alzheimer’s, Parkinson’s, multiple sclerosis), cardiovascular risk / oxidized-LDL burden, and chronic fatigue / post-viral syndromes.
Continuing Plasmalogens Week with two 2025 papers, starting with a simulated in vitro model of how humans digest mussel plasmalogens:
“Plasmalogens (Pls) have promising therapeutic potential in the treatment of neurological disorders, but their distribution, compositional intricacies, and structural alterations during the digestive process are unclear. This study aimed to address this gap by isolating Pls-enriched fractions from mussel (Mytilus edulis) and simulating their digestion in vitro across the mouth, stomach, and intestine phases.
Comparison between Pls and normal phospholipids, sharing identical fatty acyl compositions, illuminated a heightened susceptibility of Pls to catabolism during stomach digestion, which is mainly attributed to the hydrolysis reaction of Pls sensitive to acidic conditions. Phospholipid digestion commenced during the gastric phase and continued with notable catabolism in the intestinal phase, resulting in the release of substantial amounts of free fatty acids (FFAs) and lysophospholipids (LPs), which subsequently formed lipid droplets of larger sizes. Larger droplets delay intestinal absorption, extending the window period for Pls hydrolysis by pancreatic lipase.
The digestive behaviour of Pls with different polar head groups indicated that pancreatic lipase appears to digest phosphatidylethanolamine plasmalogen (PlsPE) to a greater extent than phosphatidylcholine plasmalogen (PlsPC). 41 PlsPE and 14 PlsPC were observed, suggesting that Pls may be more readily digested in the gastrointestinal tract compared to conventional phospholipids.
Generally, lipids are first absorbed by intestinal epithelial cells and undergo lipid remodeling before being transported into lymphatic fluid and then entering the bloodstream. During lipid absorption, PE can be partially converted into PC for lipid remodeling. Since in vitro digestion models cannot fully simulate the intestinal microenvironment (such as microbial metabolism and intestinal epithelial absorption), animal experiments are required to verify the actual bioavailability of PlsPE and PlsPC.”
A review highlighted nutritional implications of changes in plasmalogen chemistry:
“Plasmalogens vary quantitatively in biological systems due to biosynthesis, degradation, remodeling, and certain external stressors. Not only concentrations, but also the composition of molecular species within the plasmalogen pool changes. These shifts often involve the shortening of sn-2 fatty acyl chains, the loss of PUFAs such as DHA and EPA, and the accumulation of oxidized, truncated, or degraded species, as a result of radical-mediated oxidation and/or enzymatic degradation.
The possible increase in lysophospholipids (typically LPE and LPC, corresponding to PlsEtn and PlsCho, respectively) may be attributed to the loss of intact plasmalogens during degradation, especially in the sn-1 position. Lysoplasmalogens can be re-acylated to regenerate the original plasmalogens or create new plasmalogen species with different sn-2 fatty acyl compositions.
These molecular-level transitions highlight the complexity of plasmalogen dynamics and emphasize the need for quantitative, species-specific analysis. Variations are influenced by physiological conditions, pathological states, and nutritional supplementation.
Plasmalogens are primarily those derived from animal products, such as fish, meat, and dairy products, as well as certain marine foods. Microorganism-derived plasmalogens are attracting researchers’ attention, representing a new way of effectively utilizing bacterial resources as a ‘food’ source. Compounds provided can be plasmalogens (either PlsCho and PlsEtn, extracted from natural sources or synthesized) or plasmalogen precursors (e.g., alkylglycerols).”
A challenge researchers haven’t satisfactorily addressed yet is the question of whether beneficial oral intake of plasmalogens can be mechanistically attributed to specific plasmalogen breakdown products or to intact plasmalogens. This review introduced two other mechanistic uncertainties in that 1) absorbed and digested breakdown products can be recycled back into plasmalogens, and 2) gut microbiota can also produce plasmalogens. I’ve read papers that speculated but didn’t demonstrate that either of these factors contributed to their results.
This review cited Dr. Goodenowe’s plasmalogen precursor clinical trial mentioned in Plasmalogens Parts 1, 2, and 3. The first paper above, and most of the papers in Plasmalogen Week cited his other research.
Continuing Plasmalogens Week with two 2025 papers, starting with a rodent study of plasmalogens’ effects on mitigating cognitive decline:
“We evaluated beneficial effects of plasmalogens (PLS), phosphatidylcholine (PC), and phosphatidylserine (PS) on age-associated cognitive decline. We established a mouse model of aging-associated cognitive impairment using the subcutaneous injection of d-galactose (D-gal) at a dosage of 400 mg/kg/day.
We randomly divided six-week-old female mice into nine groups: control, model, high-dose PLS (0.3 mg/kg/day), low-dose PLS (0.09 mg/kg/day), high-dose PC (200 mg/kg/day), low-dose PC (50 mg/kg/day), high-dose PS (200 mg/kg/day), low-dose PS (50 mg/kg/day), AMC-Plas (120 mg/kg/day; and functional component PLS (0.252 mg/kg/day).
We administered PLS, PC, and PS separately by oral gavage once daily. We extracted PLS from scallops according to the literature. AMC-Plas is a commercially available health supplement known for its neuroprotective properties and memory-enhancing effects. In this study, we included AMC-Plas as a positive control group to evaluate the effects of different phospholipids.
Synaptophysin (SYP), synapsin-1 (SYN-1), postsynaptic density protein 95 (PSD-95), and brain-derived neurotrophic factor (BDNF) play important roles in synapse formation and synaptic plasticity. Synaptic function alterations or losses are key pathological mechanisms that underlie development of cognitive impairment. Therapeutic strategies that attempt to restore synaptic function or promote synaptic remodeling are considered to be increasingly promising strategies to mitigate cognitive decline.
Results showed that:
PLS improved spatial memory performance by 44% and object recognition by 80% in D-galactose-induced cognitively impaired mice.
PLS significantly decreased glial fibrillary acidic protein (GFAP)-positive cells (an indicator of astrocyte activation) in the dentate gyrus (DG) of the hippocampus, an important result because the DG is a crucial neurogenesis region.
PLS alleviated neuronal damage and protected against synaptic injury, verified by a 228% increase in PSD-95 expression in the hippocampus.
PLS showed a more prominent role for the mitigation of age-related cognitive impairment compared with PC and PS.
In conclusion, the evaluation of PLS using both behavioral and neuropathological assessments in cognitively impaired mice highlighted its exceptional efficacy compared with other phospholipids. PLS at a remarkably low effective dose significantly ameliorated cognitive deficits in cognitively impaired mice. This result further emphasized its potential relevance in neurodegenerative disease research.
We found that PLS alleviated cognitive impairment potentially by improving synaptic function; however, the molecular mechanisms that underlie its effects on synaptic function warrant further investigation.”
There was no disclosed chemical analysis of the PLS scallop extract’s plasmalogen types or other contents. Despite its name, I didn’t see that the AMC-Plas product contained plasmalogens or plasmalogen precursors.
A fruit fly study investigated plasmalogen effects on mitochondria during aging:
“We identify plasmalogens—endogenous ether-linked phospholipids—as key regulators of age-associated mitochondrial fission in Drosophila melanogaster. Loss of Kua (also known as plasmanylethanolamine desaturase (PEDS) / TMEM189 in mammals), the enzyme essential for plasmalogen biosynthesis, leads to inhibition of mitochondrial fission and impaired recruitment of the fission protein Drp1, similar to what is observed during aging.
Mitochondrial dynamics, comprising balanced cycles of fission and fusion, are essential for preserving organelle quality, metabolic flexibility, and cellular homeostasis throughout life. Aging disrupts this balance, with multiple studies reporting a decline in mitochondrial fission that contributes to the accumulation of enlarged and dysfunctional mitochondria.
These morphological changes are linked to impaired mitophagy, altered energy production, and tissue dysfunction. Midlife induction of Drp1—the dynamin-related GTPase that drives mitochondrial division—has been shown to reverse age-related mitochondrial defects and prolong lifespan in Drosophila.
To determine whether plasmalogen biosynthesis is essential for mitochondrial fission, we used KuaMI04999, a hypomorphic allele. Western blot analysis revealed significantly reduced Kua protein levels in KuaMI04999/+ heterozygotes compared to wild-type controls.
Our findings reveal a previously unrecognized lipid-based mechanism that controls mitochondrial fission during aging and position plasmalogens as key effectors linking membrane composition to mitochondrial homeostasis. It is not merely expression or stability of Drp1 that is affected, but rather its recruitment to the mitochondrial surface, which is a critical activation step for fission.
While our study highlights the requirement of plasmalogen biosynthesis for Drp1 recruitment, further work is needed to understand how plasmalogens mechanistically facilitate this interaction.”
It’s been a while since I curated plasmalogen papers. Let’s start out a week’s worth of 2025 papers with a review of plasmalogens as biomarkers:
“Reduced levels of plasmalogens in circulation or in cell membranes are associated with rare peroxisomal disorders, systemic disease, neurological impairment, cancer, and diseases of the heart, kidney, and liver. Roles for plasmalogens have been identified in lipid rafts, myelin, chlorolipids, bromolipids, hemostasis, cholesterol metabolism, and redox responses.
Plasmalogens account for approximately 5-20% of the phospholipids in mammalian cell membranes. Circulating choline and ethanolamine are incorporated into lipid membranes through the synthesis of plasmalogens. These lipids are formed through a separate multistep process involving precursors in the cytoplasm, peroxisome, and endoplasmic reticulum.
Cytochrome c (cyt-c) typically serves as an electron carrier in the mitochondrial membrane, but under oxidative stress, cyt-c undergoes a conformational alteration conferring peroxidase activity that cleaves the vinyl-ether linkage in plasmalogens. Plasmalogens may act as precursors to platelet-activating factor (PAF), and PAF can be enzymatically converted to plasmalogens. PAF is a potent pro-inflammatory mediator in cancer, cardiovascular, neurological, chronic and infectious disease, suggesting that increased PAF levels may inversely correspond to lower ethanolamine plasmalogen levels identified in human diseases.
Plasmalogens are abundant in myelin, and crucial to the function of central nervous system oligodendrocytes and peripheral nervous system Schwann cells in supporting neuronal action potential.
Catabolism of plasmalogens occurs in response to oxidative stress and activation of TLRs, which promote pro-inflammatory responses during disease progression. Release of fatty acids (e.g., arachidonic acid, eicosapentaenoic acid, docosahexaenoic acid) during plasmalogen catabolism can either exacerbate or resolve pro-inflammatory and thrombotic responses depending on the type of fatty acid released and mediator produced.
Continued research of the types of plasmalogens and plasmalogen precursors and their natural or synthetic sources, the frequency and amount of plasmalogens administered, the route of administration, and the timing of treatment is needed.”
A second review highlighted various strategies for regulating plasmalogen levels:
“Plasmalogens serve as significant structural components of cellular membranes, particularly enriched in tissues with high membrane trafficking. Plasmalogens are recognized as major reservoirs for polyunsaturated fatty acids (PUFAs), notably docosahexaenoic acid (DHA) and arachidonic acid (AA). Incorporation of these PUFAs influences membrane physical properties, including fluidity and the propensity to form non-lamellar structures.
Effective delivery of plasmalogens or their precursors faces significant hurdles, including chemical instability (especially oxidation of the vinyl-ether bond), low oral bioavailability, and challenges in crossing biological barriers like the blood–brain barrier (BBB). Exploration of plasmalogen-based nanoparticles is currently quite limited.”
A 2025 human study investigated placental and breast milk sulforaphane content:
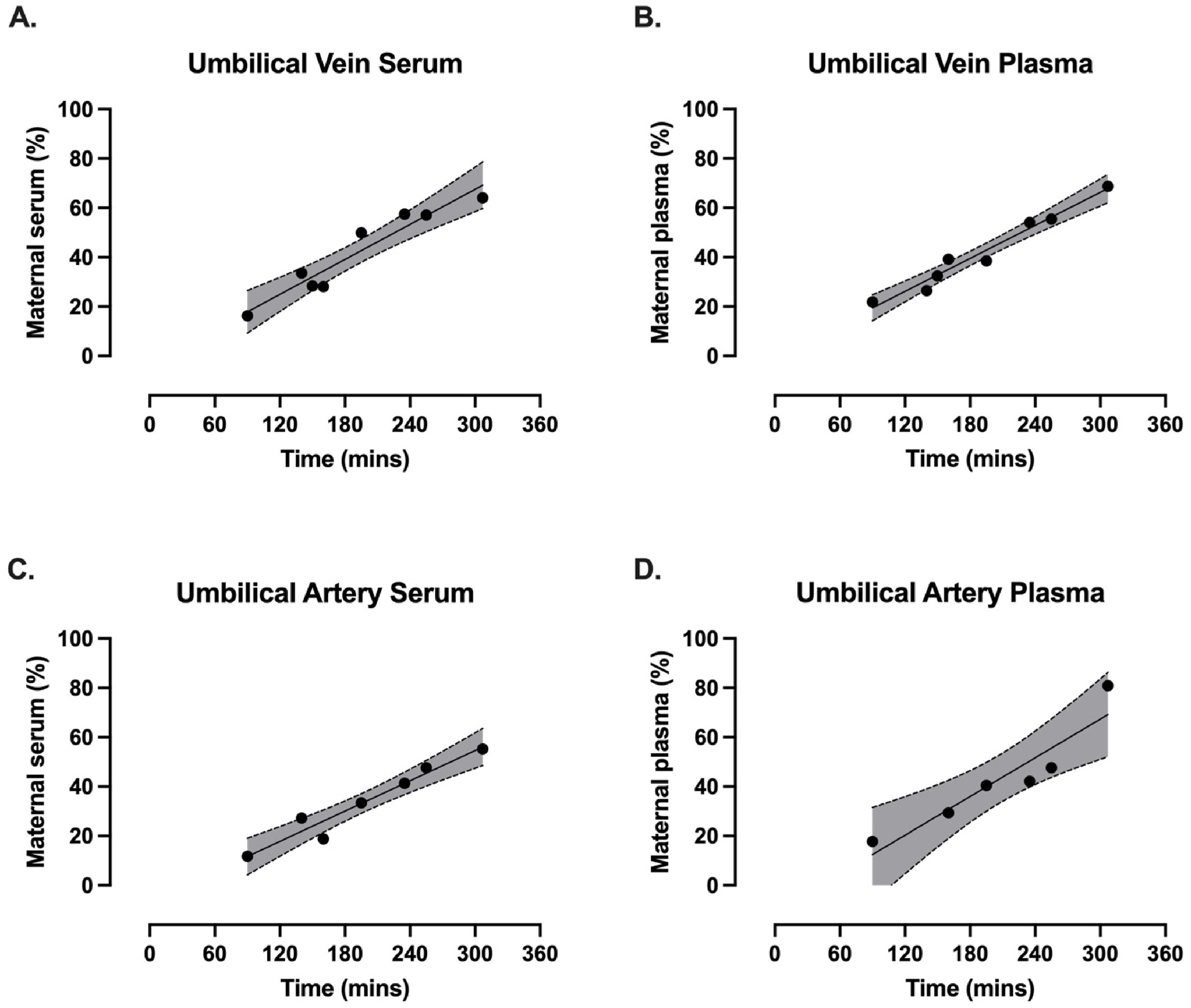
“Uncomplicated pregnant patients (n = 8) scheduled for elective caesarean sections (>37 weeks gestation) provided written and informed consent. A single oral dose of EnduraCell, a broccoli sprout extract (equivalent to 21 mg of sulforaphane), was administered prior to caesarean section. Baseline blood pressure, blood and urine were collected and again at time of operation, alongside umbilical cord blood (vein and artery) and placental samples.
2–4 days post-delivery, a second dose was administered. Two hours later, maternal bloods and breast milk were collected.
Unlike in the maternal circulation, sulforaphane levels did not show an obvious peak at the 2–3 h timepoint in the fetal umbilical vein serum and plasma or the umbilical artery serum and plasma.
A linear regression indicated that the percentage of fetal sulforaphane relative to the maternal concentration increased over time, showing progressive transfer from maternal to fetal circulation.
This is the first study to demonstrate the successful maternal-fetal transfer of sulforaphane through the placenta and into breast milk following exposure to a broccoli sprout extract during and after pregnancy. No adverse events or outcomes were reported from any of the participants, supporting the reassuring safety profile of an acute exposure to a broccoli sprout extract in pregnancy.
1. These researchers incorrectly termed a commercially available broccoli sprout powder as an extract. Grinding up broccoli sprouts produces a different product than does processing broccoli seeds or sprouts using solvents into extracts.
2. They asserted the broccoli sprout powder was a 21 mg sulforaphane dose. A more realistic explanation should have been provided, since:
No sulforaphane measurements were taken to back their assertion, which is understandable because the powder contained glucoraphanin, sulforaphane’s precursor, and sulforaphane wouldn’t be expected to be found in the powder; and
Conversion of broccoli spout powder to sulforaphane would be dependent on each subject’s gut microbiota, which is different for each individual.
“Per the manufacturer, each capsule contained 700 mg of 100% whole broccoli sprout powder, including active myrosinase and 21 mg of glucoraphanin, which upon full conversion to SFN would yield ∼8 mg, equaling ∼24 mg of SFN total per three-capsule dose. We note that full conversion to SFN, even with active myrosinase in the supplement, is not expected.”
3. Characterizing this minimal dose as “an acute exposure” mixed up its meaning with the common meaning of acute – “extremely sharp or severe; intense.”
4. Someday, researchers will be interested and forward-thinking enough about their field to plan ahead and investigate occurrences such as why both the highest and lowest maternal blood sulforaphane content didn’t translate into correspondingly ranked umbilical cord blood sulforaphane content.
5. Since blood contains up to 18,000 compounds, I don’t see where any other maternal blood compound wouldn’t pass to the fetus, unless it is definitively shown that the placenta specifically blocks it. It’s time to discard and disclaim any “safe and effective” propaganda with respect to pregnant women and breastfeeding mothers.
Except for a single intervention, therapeutic hypothermia, every non-pharmacological strategy with defined mechanisms employs more than one of these routes, most frequently pairing post-translational modification with either protein-stability regulation or limited electrophile production. This combinatorial activation elevates both NRF2 abundance and transcriptional competence while minimizing the liabilities of purely electrophilic agents and circumventing the efficacy limitations.
Classical electrophilic NRF2 activators, despite potent activation potential, exhibit paradoxically reduced therapeutic efficacy relative to single antioxidants, attributable to concurrent oxidative stress generation, glutathione depletion, mitochondrial impairment, and systemic toxicity. Although emerging non-electrophilic pharmacological activators offer therapeutic potential, their utility remains limited by bioavailability and suboptimal potency.”
https://www.mdpi.com/2076-3921/14/9/1047 “Non-Electrophilic Activation of NRF2 in Neurological Disorders: Therapeutic Promise of Non-Pharmacological Strategies”
These researchers exaggerated problems of electrophilic Nrf2 activators such as “mitochondrial impairment, and systemic toxicity” so they could have something to write about. Just like every intervention, the dose determines the response. I can’t imagine not eating broccoli sprouts in favor of brain zapping with electroconvulsive therapy or transcranial magnetic stimulation just to avoid sulforaphane’s temporary mild oxidative stress that activates Nrf2 for 15-20 minutes.
But there are limitations to how an unwell person can benefit from Nrf2 activation. For example, I haven’t curated many cancer papers because healthy body functioning can’t be assumed.
I walk the beach at sunrise, weather permitting, because it makes me feel good, and I’m always happy afterwards that I made the effort to get outside. That doing so combines two of the above non-electrophilic Nrf2 activators, physical exercise and photobiomodulation, hasn’t been a consideration.
These reviewers didn’t include human studies of sunlight’s effects. Nevermind that hospitals used to have sundecks for patients, and John Ott published relevant human and animal studies over fifty years ago.
Many studies have an undisclosed limitation in that they were performed without controlling for light. For example, knowing that mitochondria are light-activated, I don’t trust those studies’ in vivo, ex vivo, or in vitro results.
None of the 100 most recent 2025 photobiomodulation papers examined natural sunlight. Maybe it wouldn’t sell red light, green light, and blue light lasers and other products to show that people could produce the same effects themselves with sunlight at different times of the day? Would researchers damage their reputations to study a freely-available intervention, one where they don’t “do something”?
A 2025 review covered human evidence from glucosinolate and isothiocyanate research through April 2025:
“Glucosinolates (GSLs) and their breakdown products, isothiocyanates (ITCs), are biogenesis compounds with anti-inflammatory, antioxidant, and anticancer properties, mediated through key pathways such as Nrf2, NF‐κB, and epigenetic regulation. However, their limited and variable bioavailability remains a key challenge. This review summarises the current clinical evidence on GSLs and ITCs, with a focus on their health effects and metabolic fate in humans.”
https://www.mdpi.com/2304-8158/14/16/2876 “Bioavailability, Human Metabolism, and Dietary Interventions of Glucosinolates and Isothiocyanates: Critical Insights and Future Perspectives”
In the above graphic, notice how the inactive myrosinase column has no small intestine participation, but the active myrosinase column does. This point wasn’t adequately emphasized, that for complete effects, an individual has to do whatever they can to thoroughly chew or otherwise activate myrosinase to hydrolyze glucosinolates before swallowing.
Researchers don’t rely on individuals taking responsibility for their own health, of course. Just swallow these pills, we’ll do it for you, as if humans are lab rats. This lack of emphasis is understandable, if not optimal.
This review provided longish coverage of studies, which is preferable to the usual treatment of citing a reference without much explanation. Compare, for example, my longish curation of the 2023 Eat broccoli sprouts for your high intensity interval training with its reference 68 summary below:
“Another study investigated the effects of consuming GSL-rich broccoli sprout (GRS) supplements on oxidative stress and physiological adaptations to intense exercise training. In a randomised, double-blind, crossover design, nine healthy participants consumed either a GRS supplement (75 g of sprouts) or a placebo twice daily over a 7-day high-intensity interval training period. The findings revealed that GRS supplementation significantly reduced markers of oxidative stress, including carbonylated proteins in skeletal muscle and plasma myeloperoxidase levels, compared to the placebo condition. Furthermore, GRS intake led to reduced lactate accumulation during submaximal exercise and enhanced exercise performance, as indicated by a longer time to exhaustion during maximal exercise tests. At the molecular level, supplementation with GRS was associated with elevated Nrf2 protein levels in muscle tissue, suggesting activation of endogenous antioxidant defence mechanisms. In addition, GRS intake mitigated nocturnal hypoglycaemic episodes and lowered average blood glucose levels, indicating improved glucose regulation during intense training. Collectively, these results suggest that GRS supplementation may enhance physiological adaptations to high-intensity exercise by reducing oxidative stress and supporting metabolic homeostasis.”
A 2025 clinical trial compared inulin glycemic effects with FOS effects. I won’t curate its gut microbiota results as these have unresolved measurement problems:
“In this study, we conducted a randomized, double-blind investigation to examine the impact of inulin and fructooligosaccharides (FOS) on glycemic metabolism in overweight/obese and healthy adults.
Inulin and FOS are both fructans composed of fructose units, but they differ in their degree of polymerization (DP) and chain length, which lead to differences in their physicochemical properties and physiological effects. Inulin typically has a longer chain length, with a DP ≥ 10, resulting in lower solubility and slower fermentation in the distal colon. FOS consists of shorter chains, with a DP 2 to 9, presenting higher solubility and undergoing rapid fermentation in the proximal colon. These differences affect their impact on short-chain fatty acid (SCFA) production, gut microbiota modulation, and subsequently results in different effects on host metabolism.
131 participants were recruited and randomized into three groups: inulin (N = 44), FOS (N = 43), and control (N = 44). Each group was conducted with a daily supplement of 15 g FOS, inulin, and maltodextrin as placebo and lasted for 4 weeks. Dosage was determined based on our previous clinical trials in the healthy young population, which reported using 16 g/day has no risk of adverse effects. Subjects were still recommended to take a half dose in the first 2 days to promote adaptation to the prebiotics and minimize gastrointestinal symptoms. Products were suggested to add to drinks such as coffee, tea, or milk.
Inulin significantly reduced glucose levels at 1 h and 2 h during oral glucose tolerance test (OGTT), increased fasting insulin, and lowered homocysteine (HCY) levels in overweight/obese individuals. These effects were not observed in healthy individuals.
In contrast, although FOS significantly decreased HCY, it did not improve glycemic metrics in either group.”
“A 2010 gastrointestinal tolerance of chicory inulin products study indicated that 10 g/day of native inulin were well-tolerated in healthy, young adults. Over this dose would induce mild gastrointestinal symptoms.”
So a lead-in half-dose probably wouldn’t be needed for people to start a 10 gram inulin dose.
This 2025 opinion paper compared nine broccoli sprouts supplements for dogs:
“Broccoli sprouts are key elements of 9 dietary supplements for dogs. Feeding directions of 6 products correspond with consumption of dry food containing 0.5 to 29 g dried broccoli sprouts/kg. Seven supplements claim to supply sulforaphane and to possess anti-inflammatory and/or anti-cancer effects.
Directions for use of a sulforaphane-producing supplement read as follows: ‘One chewable tablet daily for dogs of all sizes, six months and older. Tablets should be given on an empty stomach at least two hours after a meal or one hour before a meal.’
There was no information found on feeding studies in dogs, addressing the impact of broccoli sprouts on health. In mice, dietary, whole-broccoli sprouts counteracted development of mammary and prostate cancer. Weights of dried broccoli sprouts in these mouse studies were 150 and 260 g/kg dry food, much higher levels than equivalents of feeding instructions for dog supplements. Species contrast and high dose blunt extrapolation of results to dogs.
The Veterinary Clinical Trials Registry of the American Veterinary Medical Association has announced that recruiting has finished for a study entitled “Sulforaphane supplementation in canine lymphoma and evaluation of epigenetic proteomic profiles”. https://veterinaryclinicaltrials.org/study/VCT17004227
The author’s use of ResearchGate is mainly to publish opinion pieces on pet animal nutrition. This doesn’t require high fees of regular journals, but also bypasses peer review.
I appreciate comparisons to rodent studies, which often intentionally overdose, and so have no relevance to humans and other mammals. His 2025 pet nutrition papers include broccoli, glyphosphate, zinc, copper, and PFAS subjects.
“Glucosinolate-rich broccoli sprouts combined with intense exercise training for 7 days have been shown to reduce blood lactate concentrations during exercise, attenuate hypoglycemic events, improve physical performance, and reduce markers of oxidative stress. This study aimed to investigate the acute, dose-dependent effects of glucosinolate-rich red kale sprouts (GRS) on blood lactate and blood glucose following the ingestion of three different doses.
Fifteen healthy participants [11 females, 4 males] consumed 37.5 g or 75 g of GRS or an isocaloric placebo blended into a beverage on three separate occasions. The participants cycled on an ergometer at three submaximal work rates before and three hours after ingestion.
Intake of glucosinolate-rich sprouts acutely decreased blood lactate levels during submaximal cycling and increased blood glucose levels at rest. The largest reduction in blood lactate was observed at the 37.5 g dose compared to placebo.
To identify the dose of GRS that results in the lowest blood lactate concentration during submaximal exercise, we applied a quadratic modeling approach. The optimal dose for minimizing lactate accumulation was calculated as 44 g of GRS.
In our previous study, we found a tendency towards a lower respiratory exchange ratio after one week of supplementation. Moreover, studies have demonstrated that mitochondrial oxidation of long-chain and short-chain fatty acids is depressed in the absence of Nrf2, and accelerated when Nrf2 is constitutively active.
We observed a reduction in myeloperoxidase levels approximately three hours after GRS intake, suggesting a decrease in oxidative stress. This finding indicates that the adaptive compensatory system may act rapidly, likely within just a few hours of GRS consumption.
A limitation is that we did not assess whether the lower lactate levels translated into improved performance. Theoretically, if the reduction in lactate results from the activation of pyruvate dehydrogenase, it could enhance performance by channeling more pyruvate into mitochondria for efficient oxidation, reducing reliance on glycolysis, and thereby sparing muscle glycogen. Alternatively, if the lower lactate levels are due to increased activity of the hepatic Cori cycle, lactate could be more rapidly converted to glucose, possibly supporting glycogen resynthesis or maintaining blood glucose levels during exercise. Both mechanisms could potentially contribute to improved performance.
Acute intake of small doses of GRS followed by submaximal ergometer cycling results in changes in lactate and glucose metabolism that could be beneficial for exercise performance.”
These researchers chose red kale sprouts of undisclosed age over the predecessor study’s broccoli raab five-day old sprouts, and two other undisclosed cruciferous vegetable sprouts.
This study is in its preprint phase. Items that could be clarified before publishing in final form include:
In the Abstract section, reference findings to red kale sprouts rather than broccoli sprouts;
Characterize the lactate U-shaped dose-response curve as hormesis; and
Two preprint studies looked at making transcriptional aging clocks using Nrf2 activators. Let’s start with a 2025 nematode study that used constant exposure to sulforaphane at different concentrations:
“To explore the potential of sulforaphane as a candidate natural compound for promoting longevity more generally, we tested the dose and age-specific effects of sulforaphane on C. elegans longevity, finding that it can extend lifespan by more than 50% at the most efficacious doses, but that treatment must be initiated early in life to be effective. We then created a novel, gene-specific, transcriptional aging clock, which demonstrated that sulforaphane-treated individuals exhibited a “transcriptional age” that was approximately four days younger than age-matched controls, representing a nearly 20% reduction in biological age.
The clearest transcriptional responses were detoxification pathways, which, together with the shape of the dose-response curve, indicates a likely hormetic response to sulforaphane. The hormetic, stress-pathway inducing properties of sulforaphane may indicate that many beneficial dietary supplements work in a fairly generic fashion as mild toxins rather than being driven by the biochemical properties of the compounds themselves (e.g., as antioxidants).
These results support the idea that robust longevity-extending interventions can act via global effects across the organism, as revealed by systems level changes in gene expression.”
There are difficulties in researchers translating nematode studies to mammals and humans. Nematodes lack a homolog to the Keap1 protein, which is sulforaphane’s main mammalian target to activate Nrf2.
A 2024 study developed various mammalian epigenetic clocks:
“A unified transcriptomic model of mortality that encompasses both aging and various models of lifespan-shortening and longevity interventions (i.e., mortality clocks) has been lacking. We conducted an RNA-seq analysis of mice subjected to 20 compound treatments in the Interventions Testing Program (ITP).
We sequenced the transcriptomes of a large cohort of ITP mice subjected to various neutral and longevity interventions, expanded the dataset with publicly available gene expression data representing organs of mice and rats across various strains and lifespan-regulating interventions, connected these models with survival data, and performed a meta-analysis of aggregated 4,539 rodent samples, which allowed us to identify multi-tissue transcriptomic signatures of aging, mortality rate, and maximum lifespan.
Aging and mortality were characterized by upregulation of genes involved in inflammation, complement cascade, apoptosis, and p53 pathway, while oxidative phosphorylation, fatty acid metabolism, and mitochondrial translation were negatively associated with mortality, both before and after adjustment for age.
Utilizing the aggregated dataset, we developed rodent multi-tissue transcriptomic clocks of chronological age, lifespan-adjusted age, and mortality. While the chronological clock could distinguish the effect of detrimental genetic and dietary models, it did not show a decrease in biological age in response to longevity interventions. In contrast, clocks of lifespan-adjusted age and mortality both captured aging-associated dynamics and correctly predicted the effect of lifespan-shortening and extending interventions.
Transcriptomic biomarkers developed in this study provide an opportunity to identify interventions promoting or counteracting molecular mechanisms of mortality, and characterize specific targets associated with their effects at the level of cell types, intracellular functional components, and individual genes. Our study underscores the complexity of aging and mortality mechanisms, the interplay between various processes involved, and the clear potential for developing therapies to extend healthspan and lifespan.”
This second study’s references included an ITP study curated in Astaxanthin and aging, which stated:
“Despite the fact that the average diet contained 1840 ppm astaxanthin (only 46% of the target), median lifespans of male UM-HET3 mice were significantly improved. Amounts of dimethyl fumarate (DMF) in the diet averaged 35% of the target dose, which may explain the absence of lifespan effects.”
So screw-ups in making both astaxanthin and DMF mouse chows ended up with study data that didn’t measure the full lifespan impacts of activating transcription factor Nrf2. I’ll assert that such faulty data may have deviated this second study by downplaying Nrf2 activation’s impact on aging, chronic disease, and rejuvenation.
Sponsors may be less likely to be presented sulforaphane and other Nrf2 activator candidates for future aging and chronic disease studies as this first study suggests, thinking that these have already been studied in mammals. Well, maybe these compounds haven’t been accurately studied. There’s no effective way to fix a rodent study’s missing DMF Nrf2 data and faulty astaxanthin Nrf2 data to train an epigenetic clock in this second study.
I could be wrong about this second study using faulty astaxanthin Nrf2 data. It was cited as Reference 27 in the Introduction as an ITP study, but not specifically cited in the Method section. I don’t know how findings such as one of Nrf2’s target genes (“Remarkably, one of the top genes positively associated with maximum lifespan and negatively associated with chronological age and expected mortality was Gpx1, encoding the selenoprotein glutathione peroxidase 1″) and a Nrf2 specific pathway (Phase II) (“Pathways positively associated with lifespan and negatively with mortality, both before and after adjustment for age, included..xenobiotic metabolism..”) were made without Reference 27. Neither of the above studies has been peer reviewed yet.