People will forgive you for being wrong, but they will never forgive you for being right – especially if events prove you right while proving them wrong. Thomas Sowell
A 2026 study investigated aging through the use of transcriptomics:
“By constructing transcriptomic clocks of expected mortality across >11,000 samples from mouse, rat, macaque and human, we provide a unified framework that integrates age-associated transcriptional change with the direction and magnitude of lifespan modulation by interventions, enabling a quantitative and biologically interpretable readout of health status.
While chronological clocks captured many detrimental conditions (e.g., chronic diseases or certain short-lived genetic models), they were comparatively less responsive to lifespan-extending interventions, consistent with reports that longevity treatments often only modestly reverse age-associated transcriptomic signatures. In contrast, mortality clocks robustly distinguished both short- and long-lived models and showed strong associations with human time-to-death, approaching the predictive performance of second-generation DNAm mortality clocks while remaining biologically interpretable.
Our results support and extend DNAm-based observations that heterochronic parabiosis in old animals and early embryogenesis can induce molecular age deceleration. In old heterochronic parabionts, mortality clocks detected a sustained tAge reduction that persisted for 2 months after detachment from young partners. Together, these findings support the view that early development contains a conserved rejuvenation-like phase, and that systemic environment can partially reset molecular age later in life.
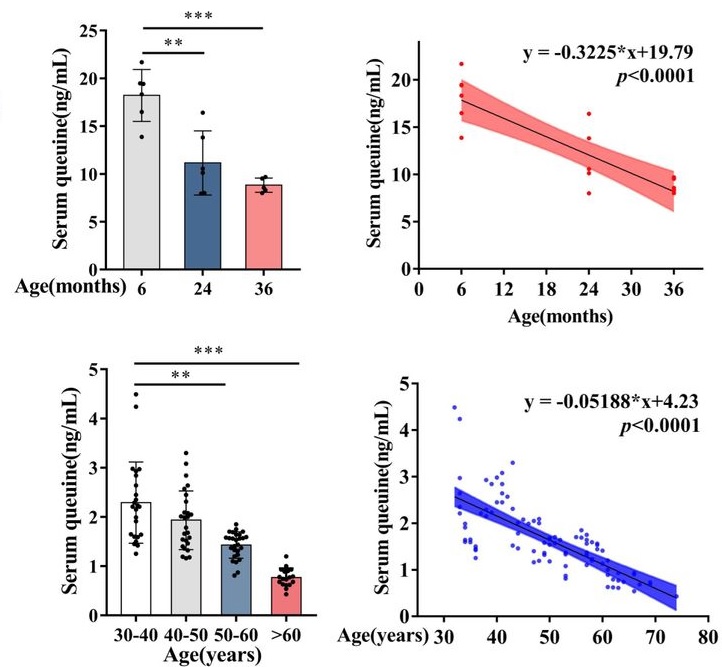
“The contribution of transfer RNA (tRNA)-specific modifications to aging remains largely unexplored. We systematically profile tRNA modifications across multiple organs, species, and senescence models, and identify mannosyl-queuosine (manQ) as the first tRNA-specific modification that consistently declines with age.
Across species, queuine supplementation extends lifespan and enhances healthspan. In naturally aging mice, long-term oral administration beginning at 16-months-old (human equivalent 50 years) extends mean lifespan by 15.3%, reduces DNA methylation age, improves cognitive and motor performance, strengthens antioxidant defenses, remodels the gut microbiota, and alleviates inflammation and metabolic dysfunction without detectable toxicity.
These findings establish tRNA epitranscriptomic remodeling as a previously unrecognized layer of aging regulation, and identify restoration of manQ through queuine supplementation as a multi-system strategy to delay aging.
manQ hypomodification is selective rather than reflecting global tRNA depletion. Aging preferentially reduces the manQ-containing tRNAAsp fragment while leaving the corresponding unmodified tRNAAsp fragment, and other queuosine-modified tRNAs, relatively unchanged.
This pattern supports a regulated defect in modification homeostasis rather than a generalized change in transcript abundance. Such specificity argues that manQ loss is not merely a passive consequence of tissue degeneration, but instead represents a conserved, biologically meaningful aging-associated event with mechanistic impact.
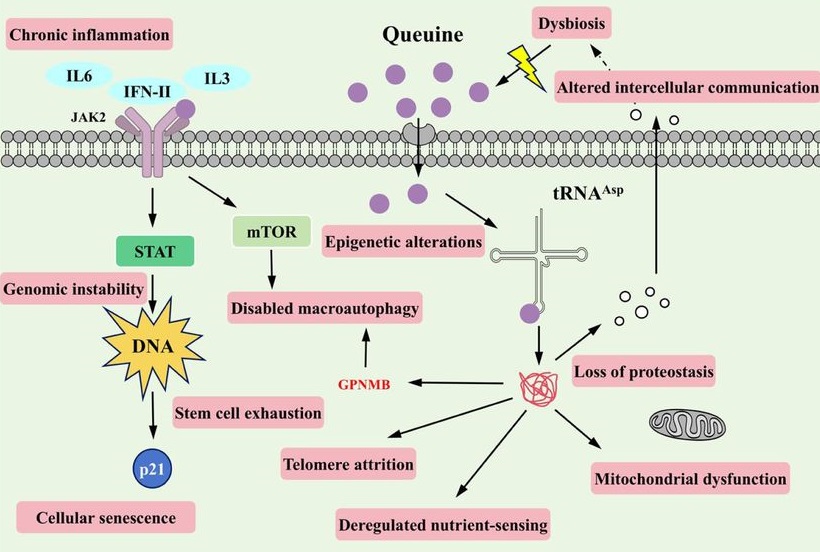
Because proteostasis intersects with multiple canonical hallmarks (e.g. mitochondrial dysfunction, impaired stress resilience, and altered intercellular communication), translation-coupled proteome destabilization offers a unifying explanation for how a single tRNA modification defect can elicit multi-system consequences. In this view, manQ decline is not merely one of many molecular changes observed in aging, but rather a proximate determinant capable of amplifying downstream hallmarks through a common axis of proteome quality control.
Our findings further suggest that manQ depletion may engage self-reinforcing feedback loops that accelerate aging trajectories. This architecture offers a conceptual framework in which aging progressively erodes ‘epitranscriptomic integrity’ at the tRNA level, pushing translation toward an error-prone regime that accelerates proteostatic collapse and functional decline.
A distinctive implication of this work is that queuine introduces a microbiota-host epitranscriptomic axis into aging biology. Queuine is produced by gut microbiota and cannot be synthesized de novo by mammals. These findings expand the conceptual scope of geroscience by placing a microbiota-derived nutrient upstream of translational quality control.
Queuine supplementation offers a distinct therapeutic logic: rather than modulating a single signaling cascade, it restores a tRNA modification state that governs translational fidelity – an upstream determinant of proteome quality that can, in principle, influence multiple downstream hallmarks concurrently. These findings highlight an intervention paradigm centered on restoring molecular fidelity, rather than suppressing a single downstream phenotype, as a strategy to delay systemic aging.”
Here are five 2025 human ergothioneine studies, starting with a clinical trial of healthy older adults:
“In this 16-week randomized, double-blind, placebo-controlled trial, 147 adults aged 55–79 with subjective memory complaints received ergothioneine (10 mg or 25 mg/day ErgoActive®) or placebo. Across all the groups, approximately 73% of participants in each group were female, with a median age of 69 years.
The primary outcome was the change in composite memory. Secondary outcomes included other cognitive domains, subjective memory and sleep quality, and blood biomarkers. At baseline, participants showed slightly above-average cognitive function (neurocognitive index median = 105), with plasma ergothioneine levels of median = 1154 nM.
Although not synthesized in the human body, ergothioneine is efficiently absorbed via the OCTN1 transporter (also known as the ergothioneine transporter, or ETT), which is expressed in many tissues, including the intestine, red blood cells, kidneys, bone marrow, immune cells, skin, and brain. This transporter enables ergothioneine to accumulate in high concentrations in organs vulnerable to oxidative stress and inflammation. Ergothioneine has multiple cellular protective functions, including scavenging reactive oxygen species, chelating redox-active metals, suppressing pro-inflammatory signaling, and protecting mitochondrial function.
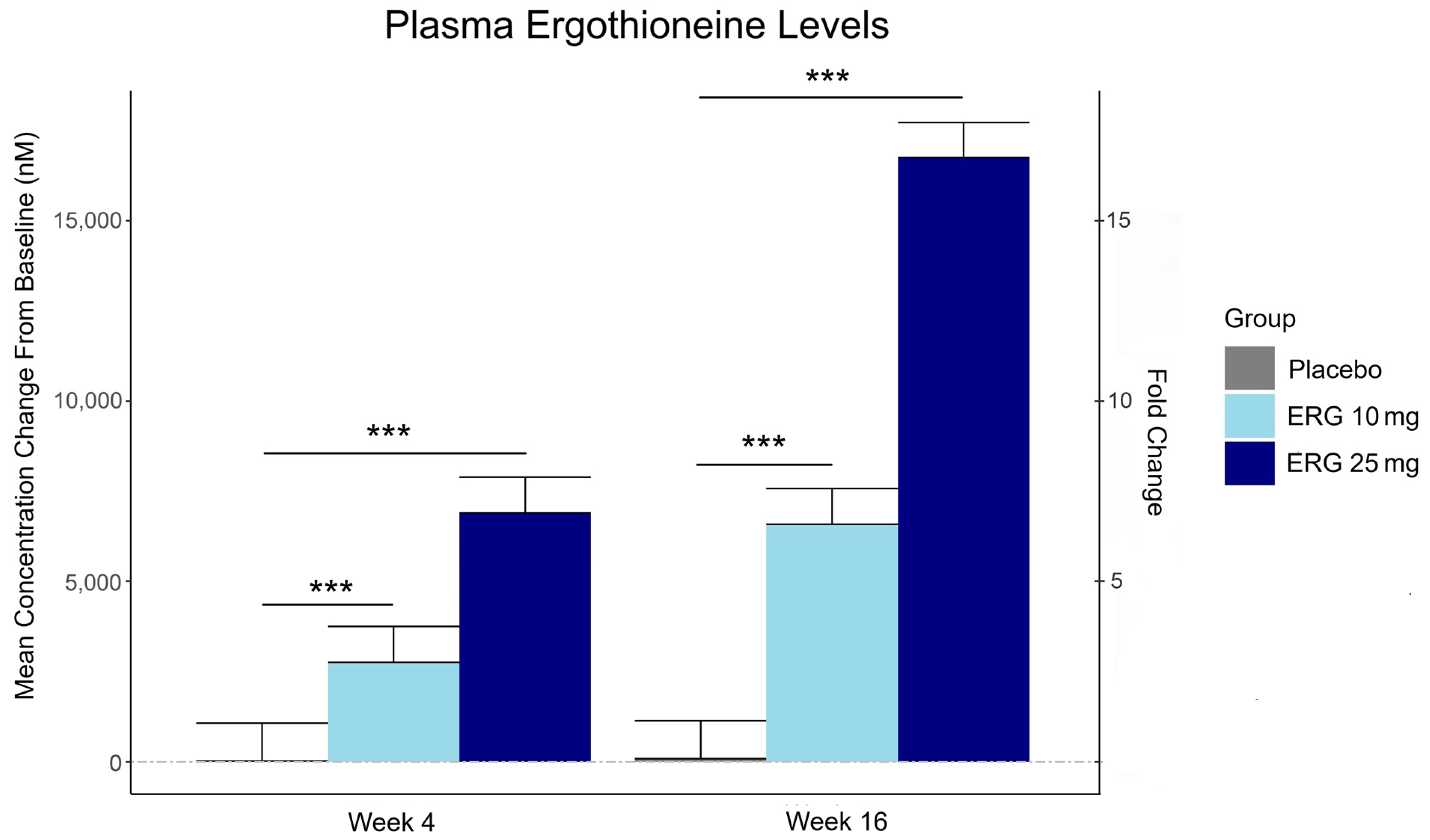
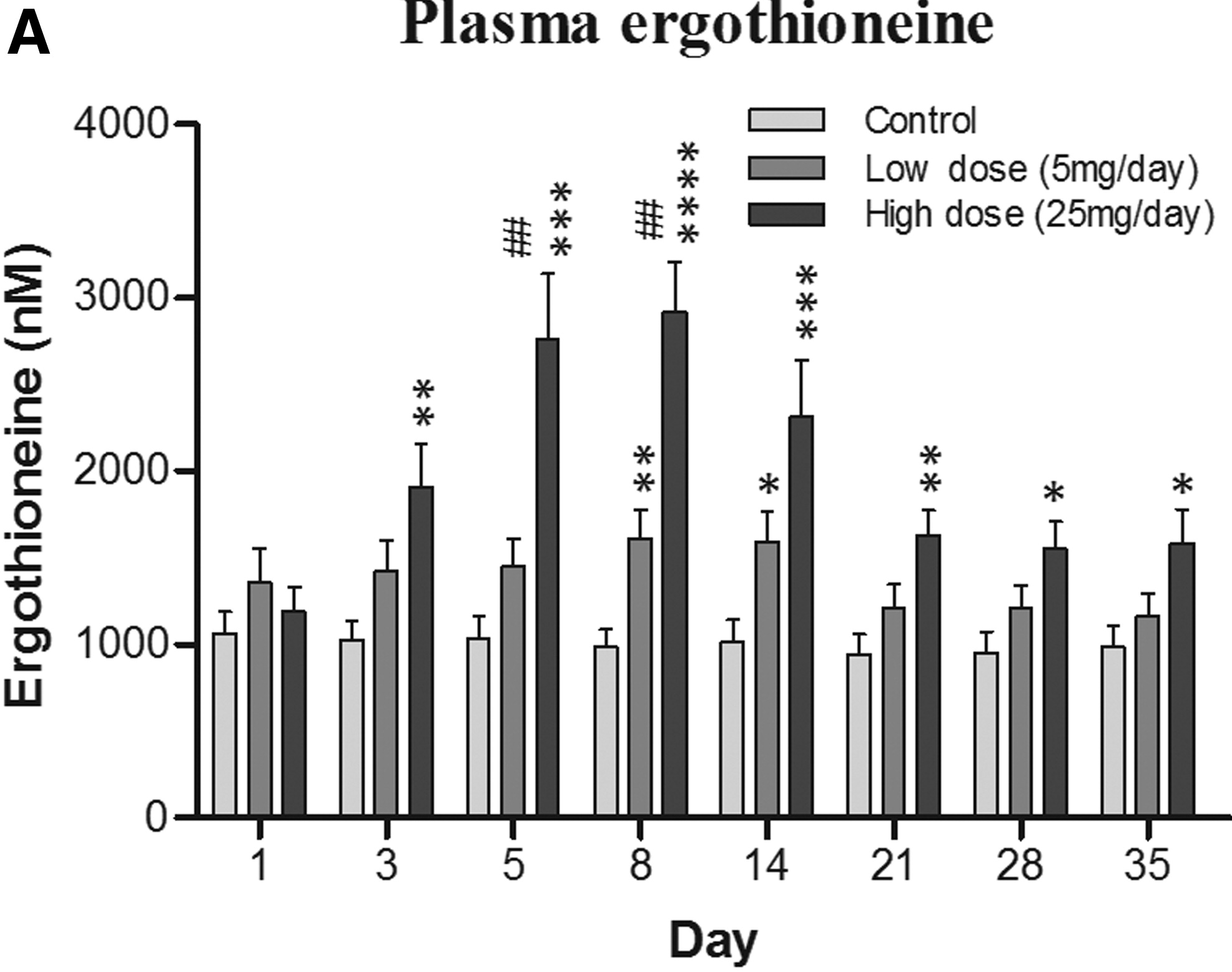
Plasma ergothioneine increased by ~3- and ~6-fold for 10 mg, and ~6- and ~16-fold for 25 mg, at weeks 4 and 16, respectively.
While the primary outcome, composite memory, showed early improvement in the 25 mg group compared to baseline, this effect was not sustained and did not differ from placebo. Reaction time showed a significant treatment-by-time interaction favoring ergothioneine, yet the between-group differences were not significant, suggesting that any potential benefits were modest and require validation in larger or longer studies.
Other cognitive effects observed were primarily within-group and not consistently dose-responsive, highlighting the challenge of detecting objective cognitive changes over a relatively short study duration in high-functioning healthy populations. However, positive effects of ergothioneine supplementation were observed on subjective measures of prospective memory and sleep initiation that were not seen in the placebo group.
This trial adds to the growing body of evidence supporting the favorable safety profile of ergothioneine. No adverse events attributable to ergothioneine were reported. Additionally, we observed potential hepatoprotective effects, with significant reductions in the plasma AST and ALT levels, particularly among males in the ERG 25 mg group.”
https://www.mdpi.com/1661-3821/5/3/15 “The Effect of Ergothioneine Supplementation on Cognitive Function, Memory, and Sleep in Older Adults with Subjective Memory Complaints: A Randomized Placebo-Controlled Trial”
The third graphic for Ergothioneine dosing, Part 2 showed a human study where a 25 mg dosing stopped after Day 7, but the plasma ergothioneine level stayed significantly higher than baseline through Day 35.
The second graphic for Ergothioneine dosing, Part 2 was a male mouse experiment where plasma ergothioneine levels of a human equivalent 22 mg to 28 mg daily dose kept rising through 92 weeks.
This trial couldn’t explain the desirability of a 25 mg daily dose that was likely (per the second and third graphics for Ergothioneine dosing, Part 2) to sustain the subjects’ increased plasma ergothioneine levels well after the trial ended at Week 16. What effects can be expected from a sustained plasma ergothioneine level that’s 16 times higher than the subjects’ initial levels? Were these 16-fold sustained plasma ergothioneine levels better or worse than the 6-fold increases in the 10 mg group, both of which were likely to continue past the trial’s end?
A representative of the trial’s sponsoring company talked a little more about the trial in this interview:
Another clinical trial investigated ergothioneine’s effects on skin:
“We conducted an 8-week, randomized, double-blind, placebo-controlled clinical trial to evaluate effects of daily oral supplementation with 30 mg of ergothioneine (DR.ERGO®) on skin parameters in healthy adult women aged 35–59 years who reported subjective signs of skin aging. Objective measurements including melanin and erythema indices, skin glossiness, elasticity, and wrinkle and pigmentation counts were used to comprehensively evaluate changes in skin condition.
The OCTN1 transporter is preferentially expressed in basal and granular epidermal layers, where cellular renewal and barrier maintenance are most active. Once internalized, ergothioneine localizes to mitochondria, where it directly scavenges reactive oxygen species (ROS) and protects mitochondrial DNA from UV- and inflammation-induced damage.
At the signaling level, ergothioneine activates key protective pathways such as the Nrf2/ARE axis, enhancing expression of antioxidant enzymes including HO-1, NQO1, and γ-GCLC. These enzymes contribute to redox homeostasis and glutathione regeneration, reinforcing cellular defense systems against photoaging and environmental insult.
In parallel, ergothioneine modulates the PI3K/Akt/Nrf2 and SIRT1/Nrf2 pathways, which are implicated in collagen preservation, inflammation resolution, and mitochondrial maintenance. These pathways converge to reduce matrix metalloproteinase (MMP) activity, enhance collagen synthesis, and suppress pro-inflammatory cytokines (TNF-α, IL-6, IL-1β), all of which are central to maintaining skin structure and function.
Compared to placebo, the DR.ERGO® ergothioneine group showed significantly greater improvements in melanin and erythema reduction, skin glossiness, elasticity, and wrinkle and spot reduction. No adverse events were reported.
These findings corroborate and extend previous clinical evidence from (Hanayama et al., 2024), who investigated an ergothioneine-rich mushroom extract (Pleurotus sp., 25 mg ergothioneine/day) in a 12-week randomized double-blind trial, and (Chunyue Zhang, 2023), who examined pure ergothioneine supplementation (25 mg/day) in a 4-week open-label study. We contextualized our results within this existing literature by comparing key outcomes.
Several limitations should be acknowledged:
The study cohort consisted solely of Japanese women aged 35–59 years, which may limit generalizability across sexes, ethnicities, and age groups.
The 8-week intervention period, while sufficient to detect short-term effects, does not allow conclusions about the sustainability of benefits or the risk of relapse upon discontinuation.
The placebo group also showed modest improvements in self-perception, highlighting the well-documented placebo response in beauty and wellness studies.
This study focused on a single daily dosage (30 mg/day) without evaluating dose–response relationships, and hydration-specific endpoints such as corneometry or transepidermal water loss (TEWL) were not included.”
Two clinical trials investigated ergothioneine’s effects on sleep quality:
“A four-week administration of 20 mg/day ergothioneine (EGT), a strong antioxidant, improves sleep quality; however, its effect at lower doses remains unclear. This study estimated the lower effective doses of EGT using a physiologically based pharmacokinetic (PBPK) model in two clinical trials.
In Study 1, participants received 5 or 10 mg/day of EGT for 8 weeks, and their plasma and blood EGT concentrations were measured. An optimized PBPK model incorporating absorption, distribution, and excretion was assembled. Our results showed that 8 mg/day of EGT for 16 weeks was optimal for attaining an effective plasma EGT concentration.
In Study 2, a randomized, double-blind, placebo-controlled study, participants received 8 mg/day EGT or a placebo for 16 weeks. The subjective sleep quality was significantly improved in the EGT group than in the placebo group.
In mammals, EGT is not generated in the body but is acquired from the diet via the carnitine/organic cation transporter OCTN1/SLC22A4. Its plasma concentration after oral administration is quite stable and gradually increases after repeated dosing on a multi-day basis.
Blood concentrations of EGT increase after Day 8 when EGT intake is interrupted, and they continue to increase until Day 35. The delayed increase in EGT concentration in the blood, compared with that in the plasma, can be interpreted as its efficient uptake by undifferentiated blood cells, which express high levels of OCTN1/SLC22A4 in the bone marrow, and subsequent differentiation to mature blood cells that enter the circulation. This may imply the nonlinear absorption, distribution, and excretion of EGT owing to saturation of the transporter at higher concentrations, potentially leading to difficulty in model construction.
This is the first study to propose a strategy to estimate lower effective doses based on the PBPK model.”
The bolded section above referenced a 2016 study / third graphic for Ergothioneine dosing, Part 2, where a 25 mg dosing stopped after Day 7, but the plasma ergothioneine level stayed high through Day 35. I didn’t see that the referenced 2004 and 2010 studies addressed this 2016 finding.
I also didn’t see that this study’s mathematical model accounted for saturation of the OCTN1 transporter or other effects, such as a very small ergothioneine clearance rate. Okay, lower the ergothioneine dose, and achieve a lower persistent plasma ergothioneine level, to what benefit?
“The present study demonstrated that OCTN1 is associated with myeloid cells rather than lymphoid cells, and especially with erythroid-lineage cells at the transition stage from immature erythroid cells to peripheral mature erythrocytes.”
Persistent high ergothioneine levels aren’t costless. Skewing bone marrow stem cells and progenitor cells toward a myeloid lineage is done at the expense of lymphocytes, T cells, B cells, and other lymphoid lineages.
Where are the studies that examine these tradeoffs? Subjective sleep quality in this study and sleep initiation in the first study above aren’t sufficiently explanatory.
A study investigated associations of plasma ergothioneine levels and cognitive changes in older adults over a two-year period:
“Observational studies have found that lower plasma levels of ergothioneine (ET) are significantly associated with higher risks of neurodegenerative diseases. However, several knowledge gaps remain:
Most of the above studies were based on cross-sectional study design, and potential reverse causation cannot be excluded. It has been suggested that plasma ET declines concomitantly with the deterioration of cognitive function.
Since the impact of a single dietary factor on health is mild, it is prone to be affected by the baseline characteristics of subjects (such as sex, educational level, disease status and gene polymorphism). However, no study has systematically evaluated potential effect modifiers on the association between ET levels and cognitive function.
The dose-response distribution between ET and cognitive function remains undetermined.
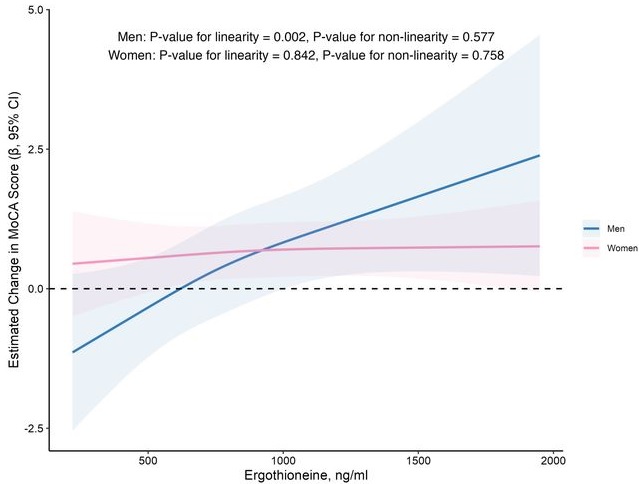
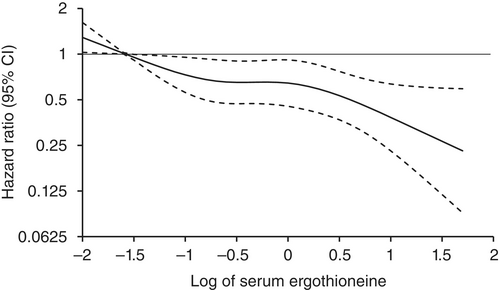
In this prospective cohort study of 1,131 community-dwelling older adults (mean age 69 years), higher baseline plasma ET levels were significantly associated with slower cognitive decline, as assessed by Montreal Cognitive Assessment (MoCA) scores, during a 2-year follow-up period.
When the plasma concentration of ET exceeds 1,000 ng/mL, the decline in cognitive function significantly slows down. However, this association has only been observed in men.
Domain-specific analysis found that the observed ET-MoCA association was mainly driven by the temporary slowdown in the decline of visuospatial/executive and delayed recall. Impaired delayed recall represents one of the earliest and most sensitive cognitive markers of dementia progression, predictive of conversion from MCI to dementia. The preferential preservation of this function by ET suggests targeted neuroprotective effects within the hippocampus.
Visual inspection of the spline curves revealed a potential plateauing effect at ET concentrations ≥1,000 ng/mL in the total population.
Baseline ET concentrations differed between men and women. Most men (81.5%) had concentrations below 1,000 ng/mL (median 754.2, IQR 592.0–937.9 ng/mL). Women exhibited substantially higher median plasma ET concentrations than men, with 35.7% of women exceeded 1,000 ng/mL (median 890.1, IQR 709.7–1,095.6 ng/mL).
Our study included only participants with normal cognitive function, and the results remained robust even after excluding those with baseline cognitive function at the lower end of the normal range. Collectively, our findings support that low ET intake occurs prior to cognitive decline.
Our findings indicate that higher plasma ET levels are significantly associated with slower cognitive decline independent of confounders in non-demented community-dwelling elderly participants, with such association observed in men but not women. Dose-response curves indicated plateauing effects above 1000 ng/mL.”
The average age of this study and the first trial above were both 69 years. Since the first trial’s participants showed slightly above-average cognitive function (neurocognitive index median = 105), with plasma ergothioneine levels of median = 1154 nM at baseline, and this study showed plateauing effects above 1000 ng/mL, I wonder how raising plasma ergothioneine levels above 1000 ng/mL could possibly show a net benefit for older people? What are the trade-offs for older people between potentially increasing slightly above-average cognitive function with ergothioneine and its other effects from saturating their OCTN1 transporter?
This study is at its preprint stage. I’m interested to see if its peer review prompts these researchers to also investigate the common finding that people who are most deficient at baseline have the greatest improvements. If so, would these sex-specific associations still hold?
Wrapping up with a study that investigated associations of serum ergothioneine levels with the risk of developing dementia:
“1344 Japanese community-residents aged 65 years and over, comprising 765 women and 579 men, without dementia at baseline were followed prospectively for a median of 11.2 years.
During follow-up, 273 participants developed all-cause dementia. Among them, 201 had Alzheimer’s disease (AD) and 72 had non-Alzheimer’s disease (non-AD) dementia.
Age- and sex-adjusted hazard ratios (HRs) for all-cause dementia, AD, and non-AD dementia decreased progressively across increasing quartiles of serum ergothioneine. These associations remained significant after adjustment for a wide range of cardiovascular, lifestyle, and dietary factors, including daily vegetable intake.
In subgroup analysis, association between serum ergothioneine levels and the risk of dementia tended to be weaker in older participants and in women:
In older individuals, cumulative burden of multiple risk factors such as hypertension, diabetes mellitus, and smoking may contribute to both neurodegenerative and vascular pathology, potentially diminishing the relative influence of ergothioneine.
In women, postmenopausal hormonal changes, particularly the decline in estrogen, have been associated with increased oxidative stress and a higher vulnerability to neurodegenerative changes.
Several limitations should be noted:
Since serum ergothioneine levels and other risk factors were measured only at baseline, we could not evaluate the changes of serum ergothioneine levels during the follow-up period. Lifestyle modifications during follow-up could have influenced serum ergothioneine levels and other risk factors. In addition, serum ergothioneine level was measured only once, and from a sample.
We cannot rule out residual confounding factors, such as other nutrients in mushrooms and socioeconomic status.
There is a possibility that dementia cases at the prodromal stage were included among participants with low serum ergothioneine levels at baseline.
We are unable to specify which mushroom varieties were predominantly consumed by participants in the town of Hisayama.
Given the limited discriminative ability of serum ergothioneine and potential degradation due to long-term sample storage, we were unable to explore a clinically meaningful threshold value of serum ergothioneine.
Generalizability of findings was limited because participants of this study were recruited from one town in Japan.
These findings suggest that the potential benefit of ergothioneine may be attenuated in individuals with pre-existing, multifactorial risk profiles for dementia.
Our findings showed that higher serum ergothioneine levels were associated with a lower risk of developing all-cause dementia, AD, and non-AD dementia in an older Japanese population. Since ergothioneine cannot be synthesized in the human body, a diet rich in ergothioneine may be beneficial in reducing the risk of dementia.”
For five years I got most of my estimated 7 mg daily ergothioneine intake from mushrooms in AGE-less chicken vegetable soup per Ergothioneine dosing. The soup was always boring, but I got too bored this year and stopped making it. I haven’t replaced mushroom intake with supplements.
I still don’t eat fried or baked foods, preferring sous vide and braising cooking methods to avoid exogenous advanced glycation end products. I avoid buying foods that evoke a hyperglycemic response or otherwise form excessive endogenous AGEs per All about AGEs.
Three papers continue Polyphenol Nrf2 activators themes starting with a 2025 review of chlorogenic acid:
“Chlorogenic acid may comprise between 70 and 350 mg per cup of coffee. Chlorogenic acid can reduce reactive oxygen species (ROS) levels via the upregulation of antioxidant enzymes, decreasing oxidative stress/damage due to the action of adaptive hormetic mechanisms. There is also a substantial literature of hormetic dose responses for metabolites of chlorogenic acid, such as caffeic acid and ferulic acid.
Chlorogenic acid-induced hormetic biphasic dose responses in a spectrum of experimental designs:
Responses to direct exposures in a range of cell types;
Preconditioning experiments in which a prior dose of chlorogenic acid protected against a subsequent stressor agent;
Studies that included direct exposure, showing hormesis dose responses and then selecting the optimal hormetic dosage as a preconditioning treatment to protect against a subsequent exposure to a toxic agent; and
A mixed group of experiments in which preconditioning was conducted, including several neuronal cellular models, all showing protection against the subsequent exposure to the toxic agent.
However, in the context of translating experimental data to clinical relevance, the concentrations employed in the majority of the in vitro studies with chlorogenic acid far exceeded transitory peak levels, even in heavy coffee drinkers (i.e., approximately 3 μM). In addition to the use of unrealistically high chlorogenic acid concentrations, exposures were prolonged, ranging from 1 to 3 days. These studies are of limited relevance to humans, a similar concern raised by other researchers involved with polyphenol research.
The present paper has framed the hypothesis that key coffee constituents, such as chlorogenic acid, show hormetic effects in a range of cell types and endpoints. Chlorogenic acid may affect some of the health benefits of coffee drinking via its role in GI tract health and beneficial brain-gut interaction.”
A 2024 review by the same research group was on hormetic effects of caffeic acid:
“Caffeic acid is a polyphenol present in numerous fruits and vegetables, especially in coffee. Diets contain about 5–10 to 50 milligrams per day of caffeic acid while coffee ingestion provides about another 250–600 milligrams per day. For the moderate to heavy coffee drinker this would result in an ingestion of about 600–1000 milligrams of caffeic acid from food and coffee consumption.
The present paper evaluates whether caffeic acid may act as an hormetic agent, mediating its chemoprotective effects as has been shown for related agents, such as rosmarinic acid, ferulic acid, and chlorogenic acid. Caffeic acid protective effects were mediated via the upregulation of a series of antioxidant enzymes related to activation of Nrf2.
Caffeic acid enhanced the lifespan of C. elegans along with similar observations for rosmarinic acid that can be hydrolyzed to caffeic acid. Several hundred plant-based agents can enhance lifespan in experimental models such as C. elegans, and there is a competition to find the most effective agents with potential commercial applications.
Hormetic effects typically show a 30 to 60% stimulation above control. This is far below the 2 to 3-fold greater than control detection limit for statistical significance based on human variability/bioplasticity and are often reported as false negatives.
A weight-of-evidence approach was proposed based on multiple in vivo and in vitro test results to derive a study design strategy to increase detection of hormetic effects within the clinical trial framework. Such research should explore hormetic based interactions linking protective catabolic-based adaptive responses with activation and regulation of anabolic mediated hormetic growth effects.”
A 2024 review provided an overall picture of coffee compounds’ cardiometabolic effects:
“This review provides a comprehensive synthesis of longitudinal observational and interventional studies on the cardiometabolic effects of coffee consumption.
Findings indicate that while coffee may cause short-term increases in blood pressure, it does not contribute to long-term hypertension risk.
There is limited evidence indicating that coffee intake might reduce the risk of metabolic syndrome and non-alcoholic fatty liver disease.
Coffee consumption is consistently linked with reduced risks of type 2 diabetes (T2D) and chronic kidney disease (CKD), showing dose-response relationships.
The relationship between coffee and cardiovascular disease is complex, showing potential stroke prevention benefits but ambiguous effects on coronary heart disease.
Moderate coffee consumption, typically ranging from 1 to 5 cups per day, is linked to a reduced risk of heart failure, while its impact on atrial fibrillation remains inconclusive. Coffee consumption is associated with a lower risk of all-cause mortality, following a U-shaped pattern, with the largest risk reduction observed at moderate consumption levels.
Except for T2D and CKD, Mendelian randomization studies do not robustly support a causal link between coffee consumption and adverse cardiometabolic outcomes.
Potential beneficial effects of coffee on cardiometabolic health are consistent across age, sex, geographical regions, and coffee subtypes and are multi-dimensional, involving antioxidative, anti-inflammatory, lipid-modulating, insulin-sensitizing, and thermogenic effects. Based on its beneficial effects on cardiometabolic health and fundamental biological processes involved in aging, moderate coffee consumption has the potential to contribute to extending healthspan and increasing longevity.”
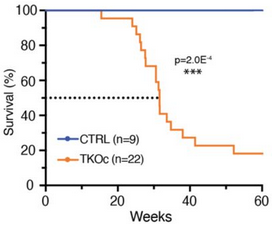
A 2024 rodent study investigated epigenetic effects of loosening compacted chromatin:
“We show using a novel mouse strain, (TKOc), carrying a triple knockout of three methyltransferases responsible for H3K9me3 deposition, that the inducible loss of H3K9me3 in adulthood results in premature aging. TKOc mice exhibit:
Reduced lifespan;
Lower body weight;
Increased frailty index;
Multi-organ degeneration;
Transcriptional changes with significant upregulation of transposable elements; and
Accelerated epigenetic age.
Through simultaneous depletion of Setdb1 and Suv39h1/2 methyltransferases, crucial to formation of constitutive heterochromatin, our model analyzes consequential transcription changes including a potential source of genomic instability by activation of endogenous mobile genetic elements, specifically transposable elements.
These findings reveal the importance of epigenetic regulation in aging, and suggest that interventions targeting epigenetic modifications could potentially slow down or reverse age-related decline.”
Many of these findings could be restated without viewing them as age-related, i.e.: failure to maintain an adult’s methyltransferase system results in a loss of health. For example, an unhealthy methyltransferase system indicated by parameters like homocysteine levels (not mentioned) can be reversed to healthy function regardless of age. Healthy vs. unhealthy system function wasn’t the paradigm these researchers operated under, though.
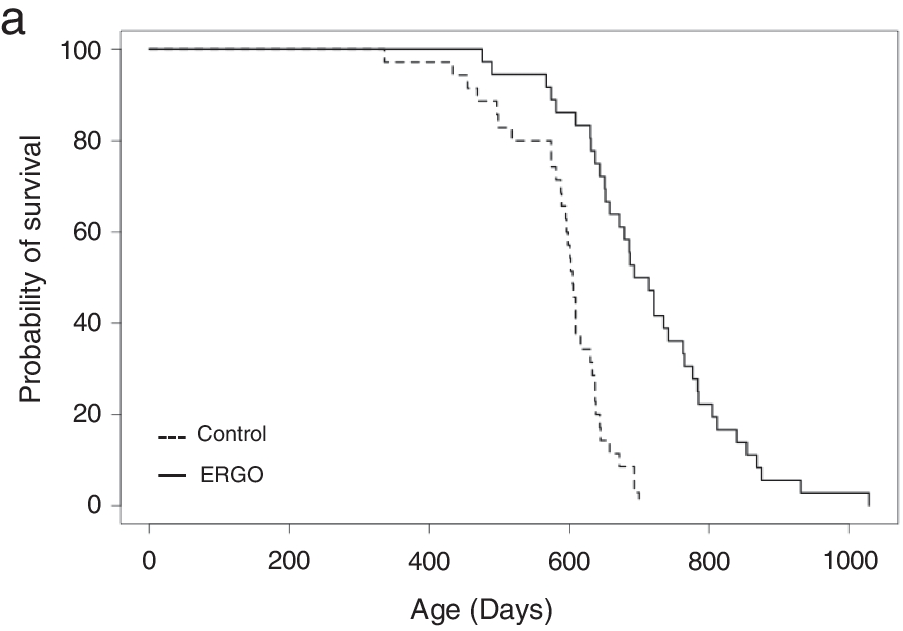
Continuing Part 1 with a 2024 rodent healthspan and lifespan study:
“We investigated the effects of daily oral supplementation of ergothioneine (ERGO) dissolved in drinking water on lifespan, frailty, and cognitive impairment in male mice from 7 weeks of age to the end of their lives. Ingestion of 4 ~ 5 mg/kg/day of ERGO remarkably extended the lifespan of male mice.
The ERGO group showed significantly lower age-related declines in weight, fat mass, and average and maximum movement velocities at 88 weeks of age. This was compatible with dramatic suppression by ERGO of age-related increments in plasma biomarkers. ERGO also rescued age-related impairments in learning and memory ability.
Ingestion of ERGO may promote longevity and healthy aging in male mice, possibly through multiple biological mechanisms.”
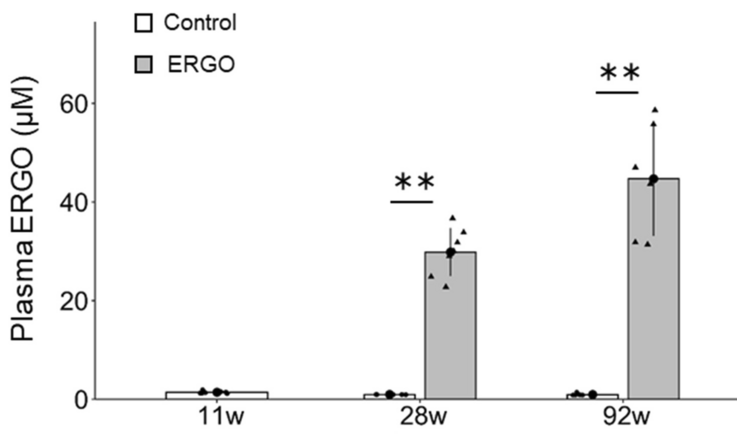
Subjects’ plasma ergothioneine levels of an estimated 4 ~ 5 mg/kg daily dose were:
A human equivalent daily dose is an estimated 22 mg to 28 mg (4 or 5 mg x .081 x 70 kg).
The third paper in Part 1 cited a 2017 clinical trial that provided 5 mg and 25 mg ergothioneine doses for 7 days, resulting in these plasma ergothioneine levels:
The first paper of Part 1 referenced a 2020 human study where the dose was 5 mg/day for 12 weeks, but I don’t have access to it. It’s unclear whether humans could continually raise ergothioneine levels by daily consumption throughout our lives as did this rodent study.
A 2024 paper reviewed the importance of ergothioneine to humans:
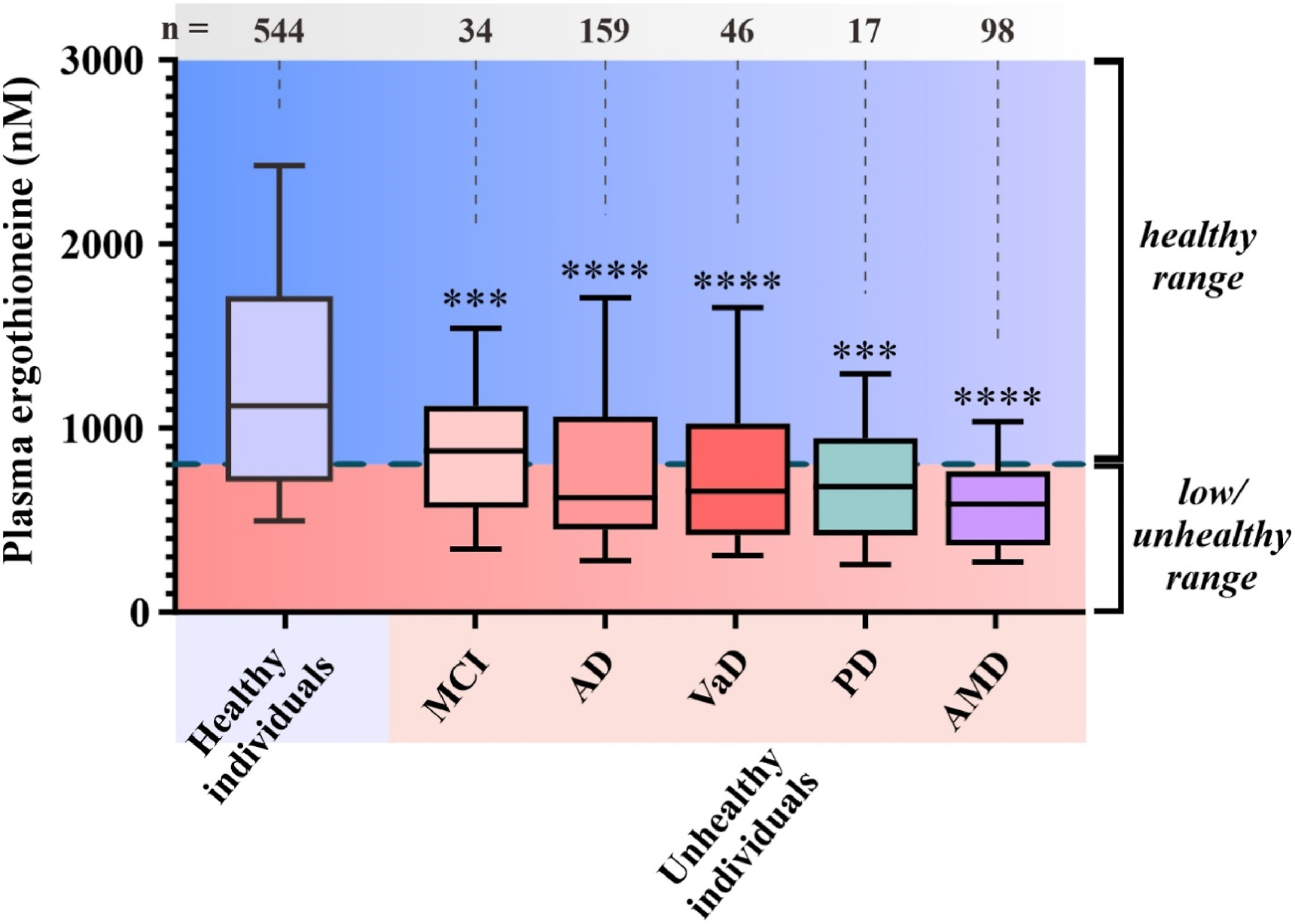
“We propose that the diet-derived compound ergothioneine (ET) is an important nutrient in the human body, especially for maintenance of normal brain function, and that low body ET levels predispose humans to significantly increased risks of neurodegenerative and possibly other age-related diseases.
Work by multiple groups has established that low ET levels in humans are associated not only with cognitive impairment/AD but also with other age-related conditions, including frailty, Parkinson’s disease, vascular dementia, chronic renal disease, cardiovascular disease, and macular degeneration. Low ET levels also correlate with increased risk of developing preeclampsia in pregnant women [53].
Plasma ET levels from healthy (age-matched) vs unhealthy individuals in Singapore – Mild cognitive impairment (MCI); Alzheimer’s disease (AD); vascular dementia (VaD); Parkinson’s disease (PD); age-related macular degeneration (AMD):
Does low ET cause or contribute to age-related neurodegeneration, or
Does disease cause low ET, or
Low ET and increased disease risk are both caused by something else, as yet unidentified?
Prevention of neurodegeneration is especially important, since by the time dementia is usually diagnosed damage to the brain is extensive and likely irreversible.”
Whether or not the healthy individuals ate mushrooms daily in the above graphic was lost while conglomerating multiple studies.
Note that scales of the above two human graphics are a thousand times smaller than the above rodent graphic. I thought that maybe the rodent study made a plasma ergothioneine calculation error, but didn’t see one in the provided Supplementary data.
Reference 53 of the second paper is a 2023 human study:
“We analysed early pregnancy samples from a cohort of 432 first time mothers. Of these 432 women, 97 went on to develop pre-term or term pre-eclampsia (PE).
If a threshold was set at the 90th percentile of the reference range in the control population (≥462 ng/ml), only one of these 97 women (1%) developed PE, versus 96/397 (24.2%) whose ergothioneine level was below this threshold. One possible interpretation of these findings, consistent with previous experiments in a reduced uterine perfusion model in rats, is that ergothioneine may indeed prove protective against PE in humans.”
Eyeballing the Healthy individuals in the above graphic, none of those 544 people were below this study’s 462 ng threshold.
A 2023 companion article analyzed the third paper’s unusual findings:
“These results suggest that there might be a dichotomized association between ergothioneine concentrations and preeclampsia; and only a high ergothioneine level over 90th percentile of the control population could be protective against preeclampsia.
Univariable results showed that ergothioneine had a significant non-linear association with preeclampsia and it would start to offer protective effect from 300 ng/ml onward. Analysis also confirmed that body mass index was significantly associated with an increased risk of preeclampsia.
A large observational study could strengthen the causal association between ergothioneine and preeclampsia. If confirmed, a randomized controlled trial (RCT) assessing whether ergothioneine supplementation can reduce risk of preeclampsia will be imminently feasible. Ideally, such RCT should compare placebo with a range of different doses of ergothioneine to identify the best or minimal effective dose, given its good safety records, including in pregnancy, with a no-observed-adverse-effect level (NOAEL) of 800 mg/kg body weight per day.”
My daily mushroom ergothioneine dose is around 7 mg, and I weigh about 70 kg. I don’t think a daily 800 mg/kg ergothioneine dose would be desirable for anybody, regardless of what experts say.
How many times have public health employees been wrong this decade? Would you bet your or your child’s health on their advice?
“Pronounced rejuvenation effects in male rats prompted us to conduct further confirmatory experiments. A particularly important consideration is the effectiveness of E5 with regards to sex, as sex-dependent rejuvenation by some interventions have previously been reported.
To assess E5’s applicability to both male and female Sprague Dawley rats, we studied 12 males (6 treated with E5, 6 with saline) and 12 females (6 treated with E5, 6 with saline). These rats were treated every 45 days with an injection of E5 or saline. Rats were monitored for 165 days, and blood was drawn at six time points: 0, 15, 30, 60, 150 and 165 days from the first injection.
We observed highly significant improvements in TNF alpha and IL-6 levels for both males and females in the blood of E5-injected rats over that of saline controls. We also observed a substantial improvement in grip strength.
Our study shows age reversal effects in both male and female rats, but E5 is more effective in males.”
Another experimental group was started with old rats of both sexes. Using the human / rat relative clock developed in the original study, a human equivalent age to these rats at 26 months old was ((112.7 weeks / 197.6 weeks maximum rat lifespan) x 122.5 years maximum human lifespan) = 69.8 years:
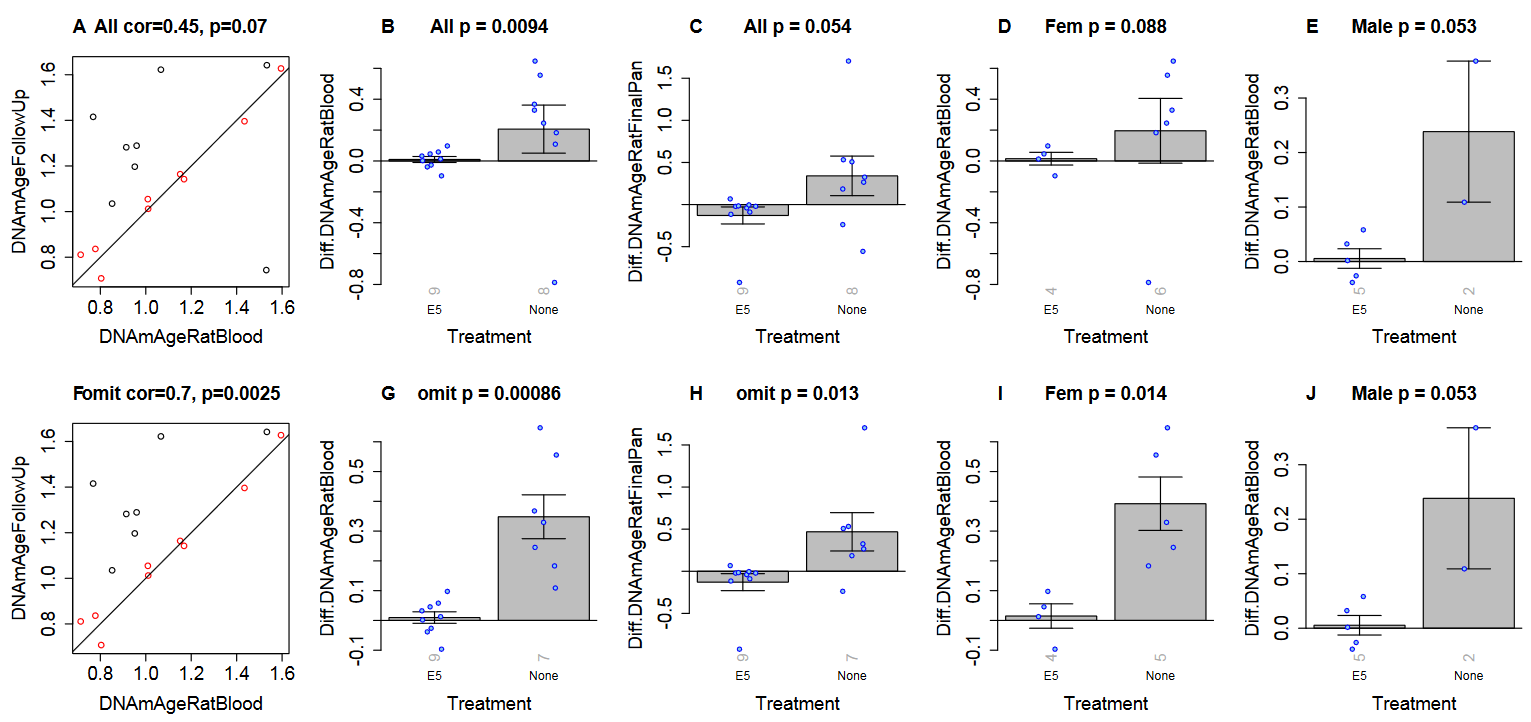
“To validate our epigenetic clock results, we conducted a second set of E5 experiments with Sprague Dawley rats of both sexes. When these rats turned 26 months old, half (9 rats) received the E5 treatment while the other half (8 rats) received only the control treatment (saline injection). We analyzed methylation data from two blood draws: blood draw before treatment (baseline) and a follow up sample (15 days after the E5/saline treatment).”
Treatment measurements were affected by one female control group outlier. Panels F through J were recalculated after removing the outlier to show significant effects in both sexes:
“A) Final version of the rat clock for blood. Baseline measurement (x-axis) versus follow up measurement (15 days after treatment, y-axis). Points (rats) are colored by treatment: red=treated by E5, black=treated with saline only. Rotated grey numbers underneath each bar reports the group sizes. Each bar plot reports the mean value and one standard error.
B,D,E) Difference between follow up measurement and baseline measurement (y-axis) versus treatment status in B) all rats, D) female rats only, E) male rats only. C) is analogous to B) but uses the pan tissue clock for rats.
Panels in the second row (F,G,H,I,J) are analogous to those in the first row but the analysis omitted one control rat (corresponding to the black dot in the lower right of panel A).”
A description of how E5 plasma fraction was made starts on page 16 of the *.pdf file. The next E5 study will be done with dogs per July 2023 updates in blog post comments:
“On E5 our entire team is working hard towards the launch of an old Beagle dogs trial this month. We want to make them really young, healthy, happy, and jumping around like 1 and 2 year olds.
Primary endpoint is safety and toxicology to test various dose strengths and frequencies. Secondary endpoints are more than 20.
As you know, we like to test exhaustively to get a sharper perspective of what’s happening. In rat studies we tested 30 biomarkers, including functional. We are especially keen to check kidney markers.
There are two clocks for dogs we are interested in to get third party confirmation of age reversal. Horvath dog clock is ready and GlycanAge dog clock is under construction.
We are requesting all organizations that support pets and aging to financially support their project of building an accurate dog clock. Not only will it help veterinary aging research like ours, but also all the dog owners that may want to know how much improvement their dog received from treatment. Dr. Matt Kaeberlain is an advisor on their project.”
An August 2023 interview with Dr. Dale Bredesen, who has reversed Alheizmer’s disease in many people, which will never be acknowledged by the corrupt paradigm:
“How much do you want me to go into things that are relatively controversial and how much do you want me to stick with kind of the more standard line?
For Alzheimer’s we noticed initially there are 36 different potential contributors. You need to patch as many as possible to have an effect.
All of these things, your estradiol level, your progesterone level, pregnenolone, free T3, TSH, Vitamin D, testosterone, these things are all critical. They all feed into the equation.
You have over a hundred trillion contacts in your brain. Will you be able to keep them? Or do you not have what it takes to keep them, and you have to downsize?
The reality is Alzheimer’s disease should be a rare disease. If everybody would get on appropriate prevention or early reversal, we could make it a rare disease.”
A 2023 human / primate / rodent / worm study with 56 coauthors exhaustively investigated taurine effects:
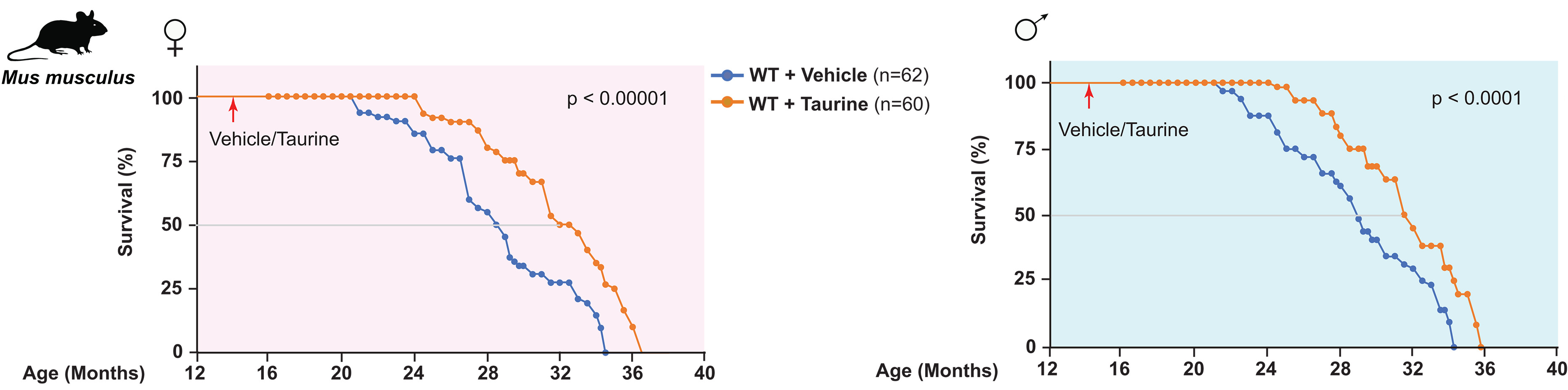
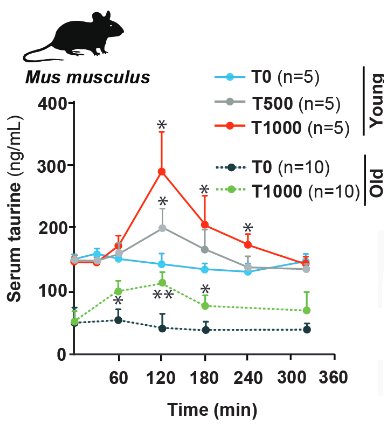
“We measured the blood concentration of taurine during aging and investigated the effect of taurine supplementation on healthspan and lifespan in several species.
In C57Bl/6J wild-type (WT) mice, serum taurine concentrations declined from 132.3 ± 14.2 ng/ml at 4 weeks to 40.2 ± 7.1 ng/ml at 56 weeks.
In 15-year-old monkeys, serum taurine concentrations were 85% lower than in 5-year-old monkeys.
Taurine concentrations in elderly humans were decreased by more than 80% compared with concentration in serum of younger individuals.
Regardless of their sex, taurine-fed mice survived longer than control mice. The median lifespan increase was 10 to 12%, and life expectancy at 28 months increased by 18 to 25%.
Improved survival of taurine-fed mice was not a consequence of low survival of control animals or differences in diet. Taurine deficiency is a driver of aging in mice because its reversal increases lifespan.
We investigated the health of taurine-fed middle-aged mice and found an improved functioning of bone, muscle, pancreas, brain, fat, gut, and immune system, indicating an overall increase in healthspan. Taurine reduced cellular senescence, protected against telomerase deficiency, suppressed mitochondrial dysfunction, decreased DNA damage, and attenuated inflammation.
An association analysis of metabolite clinical risk factors in humans showed that lower taurine, hypotaurine, and N-acetyltaurine concentrations were associated with adverse health, such as increased abdominal obesity, hypertension, inflammation, and prevalence of type 2 diabetes. We found that a bout of exercise increased concentrations of taurine metabolites in blood, which might partially underlie antiaging effects of exercise.
Taurine abundance decreases during aging. A reversal of this decline through taurine supplementation increases healthspan and lifespan in mice and worms, and healthspan in monkeys.”
“One of the most studied mechanisms of action for taurine is an increase in antioxidant capacity. Although oxidative damage is not clearly linked to mammalian lifespan, it plays a role in many age-associated pathologies.
Taurine is a poor scavenger of reactive oxygen species, with the exception of hypochlorite, which it detoxifies to N-chlorotaurine. N-Chlorotaurine is anti-inflammatory and induces expression of antioxidant enzymes in mice and humans.
Taurine supplementation might also cause an increase in levels of its precursors, including the antioxidants hypotaurine and cysteine. An interesting corollary is that up-regulating endogenous taurine synthesis would have the opposite result—consuming hypotaurine and cysteine.”
A human equivalent taurine dose is (1 g x .081) x 70 kg = 5.67 grams. Dose tests from supplementary data were:
“Dose and frequency of taurine administration was selected based on a pilot study, which showed that when given once daily to middle-aged WT mice, this regimen increased peak blood taurine concentrations to baseline concentrations in young (4-week-old) mice.”
I’ve taken 2 grams every day for the past three years, and will now bump that up to 5 grams. My diet doesn’t regularly include any foods high in taurine.
I recommend reading the study rather than commentaries. Its publisher did a very good job of linking figures so that images can be viewed, then the reader returned to the right context.
Gatekeepers are out in full force on this study, and their viewpoints are probably what you’ll see first, to include unevidenced statements like “the study’s main authors cautioned the public not to self-dose with the supplement” and the above introductory article’s unreferenced “equivalent doses used in the study by Singh et al. would be very high in humans.” Pretty pathetic that such ‘authorities’ are even publicized after recent years of deliberately misleading the world about science and medicine.
This study and all commentaries called for clinical trials that are NOT going to happen:
Drug companies can’t make money from a research area that’s cheap, not patentable, and readily accessible.
Government sponsors are likewise not incentivized to act in the public’s interest per their recent behavior.
Take responsibility for your own one precious life. See Part 2 for a sample of citing papers.
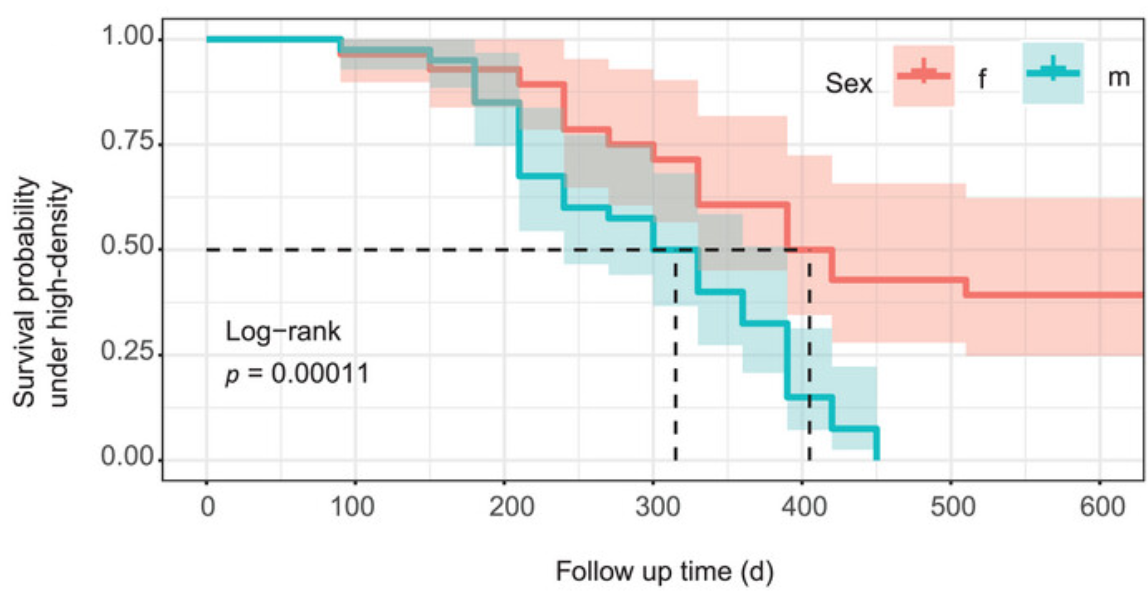
This 2023 rodent study investigated aging processes and gut microbiota in crowded conditions:
“Our study provides clear evidence that high-density crowding accelerates the aging process of Brandt’s voles. We also found that ‘high-density microbiota’ promote the aging-related phenotype in voles.
Because we minimized effects of direct fighting on mortality of voles, observed changes in lifespan in this study should mostly represent the natural aging processes of voles.
High density increased the level of stress hormone corticosterone, which disrupted gut microbiota composition by:
Decreasing abundance of anti-aging or anti-inflammatory bacterial species; and
Increasing the proportion of pathogenic bacteria.
This caused an increase in DNA oxidation and inflammation through upregulation of NF-kB and COX-2 pathways.
Although high-density relief and butyric acid administration interventions could reverse aging-related processes of adult voles, it remains unclear whether they could reverse the aging process in terms of lifespan.
Our results suggest that gut microbiota play a significant role in mediating aging-related processes of voles under high-density conditions, and can be used as a potential therapeutic target for treating stress-related diseases in humans.”
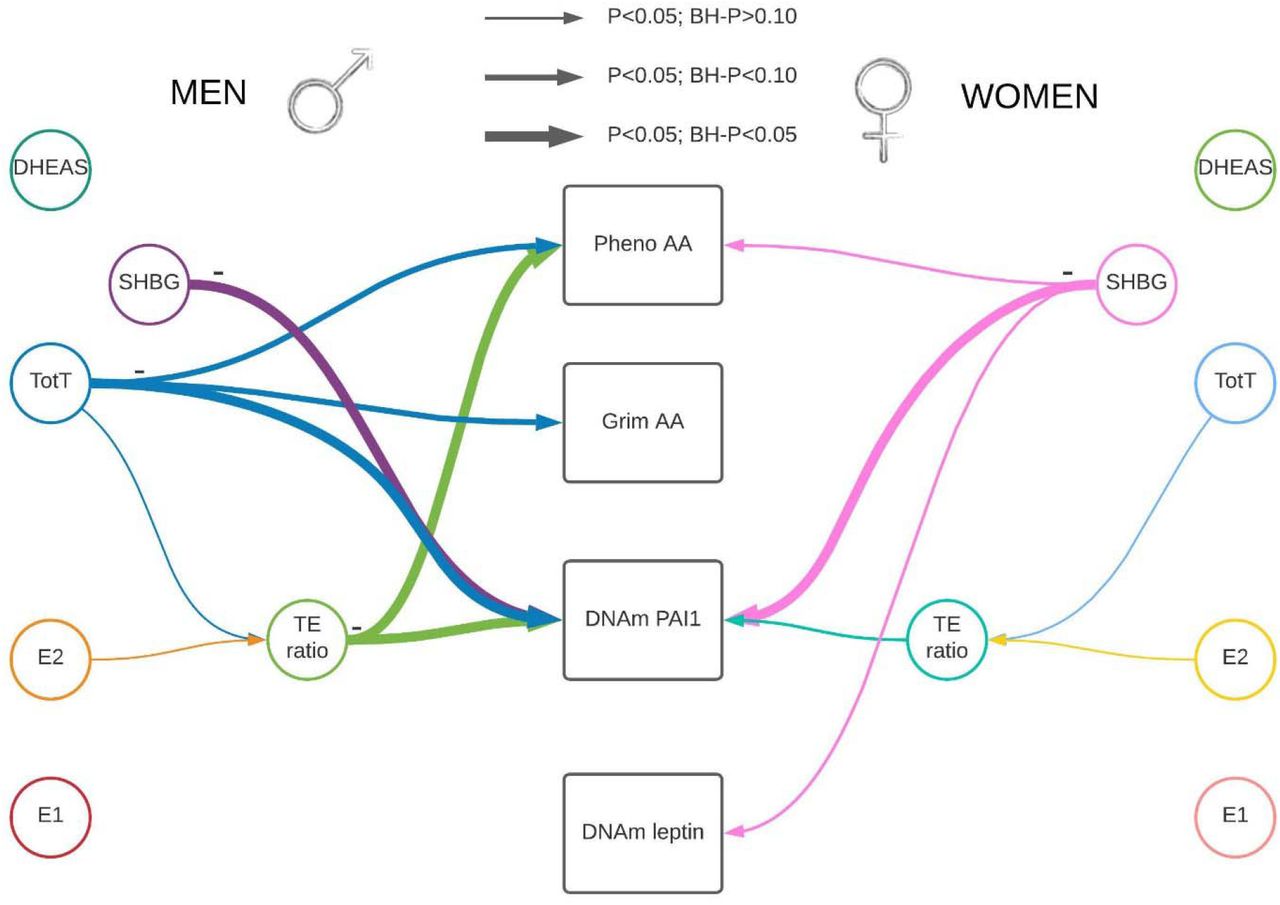
This 2023 human study investigated associations among sex hormones and epigenetic clocks:
“We studied associations between sex steroid hormones and DNA methylation-based (DNAm) biomarkers of age and mortality risk including Pheno Age Acceleration (AA), Grim AA, and DNAm-based estimators of Plasminogen Activator Inhibitor 1 (PAI1), and leptin concentrations.
Leptin is a peptide hormone and is associated with regulation of food intake and energy balance. Leptin also influences inflammatory processes, angiogenesis, lipolysis, and neuroplasticity.
PAI1 is a protein that is involved in tissue hemostasis. Previous studies that assessed associations between sex hormones and PAI1 protein concentrations in blood reported conflicting results.
DNAm PAI-1 was shown to be a better surrogate for lifespan than the actual plasma measure, and performs better than Grim AA regarding associations with the comorbidity-index. Another potential benefit of using DNAm-based biomarkers instead of plasma biomarkers is that the DNAm-based biomarkers represent a longer average estimate of biomarker concentration, and are not as affected by day-to-day variations that could bias results.
Associations are represented by colored arrows with the lines’ thickness representing association strength. As the association was measured mainly cross-sectional, association directionality cannot be established.
Hormone levels were inversely associated with epigenetic estimators of mortality risk.
Sex Hormone Binding Globulin (SHBG) was associated with a decrease in DNAm PAI1 among men and women.
Higher testosterone and testosterone/estradiol ratio (TE) were associated with lower DNAm PAI and a younger epigenetic age in men.
A decrease in DNAm PAI1 is associated with lower mortality and morbidity risk indicating a potential protective effect of testosterone on lifespan and conceivably cardiovascular health via DNAm PAI1.”
This 2022 human study investigated another type of aging clock:
“The glycan clock of age, based entirely on immunoglobulin G (IgG) N-glycans, can predict biological age with high accuracy. Unlike DNA methylation, glycosylation of IgG does not predict chronological age with high accuracy.
Heritability analysis of plasma glycans revealed that the majority of traits have high heritability estimates, indicating a tight genetic control of glycosylation. To better understand genetic and environmental factors influencing glycan clock variation, we performed a heritability analysis on data from two cohorts included in the TwinsUK registry.
Glycosylation is a series of enzymatic reactions in which carbohydrates are attached to other molecules (e.g., proteins or lipids) resulting in formation of complex carbohydrates and glycoconjugates commonly referred to as ‘glycans.’ Glycosylation of IgG antibody is especially interesting as it dramatically affects its function, and acts as a molecular switch between pro- and anti-inflammatory immune responses.
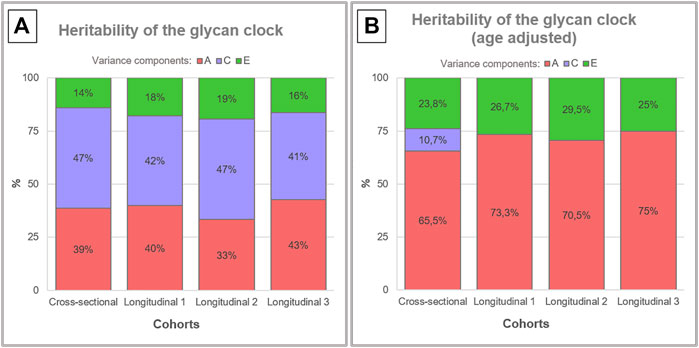
Heritability of the glycan clock was estimated to decompose observed phenotypic variance into three latent sources of variation:
A—additive genetic variance [red] represents cumulative impact of genes;
C—shared/common environment variance [purple] results from influences to which both members of a twin pair are exposed; and
E—unique environment variance [green] is events occurring to one twin but not the other, and includes measurement error.
Despite tight genetic control of the IgG glycome:
Heritability analysis of the glycan clock revealed only a moderate genetic contribution averaging around 39% [A, left side].
Including age of the individuals as a covariate in heritability analysis averaged 71% heritability estimates [B, right side].
Mean time difference was 7.5 years for points 1 and 2, and 6 years for points 2 and 3.
Observed increase in the genetic component could be a consequence of chronological age as a shared environmental variance characteristic for every individual and determined by their genetic makeup and epigenetic regulation.”
Although A rejuvenation therapy and sulforaphane was cited, these researchers missed its central premise: Pro-aging epigenetic programming is directional and not purely random. Contrasting their above graphic’s heritability estimates of 39% with the age-regressed, right side’s average 71% could hardly have been more clear in illustrating this fact.
This study instead stated “Aging in general leads to epigenetic mediated deregulation of genes.” This weak sauce accompanied speculations such as “supports the notion that the glycan clock can be rejuvenated by simple lifestyle choices.”
Researchers almost always want to claim being first in finding x, y, or z. These researchers could have done that in this glycan clock study by highlighting an outstanding finding. So what happened?
An alternate explanation to their paradigm blinding them could be sponsor expectations, peer pressures, etc. I’ll ask them about it, and will update here with their response.
Five 2022 papers focusing on walnuts, starting with a comparison of eight tree nuts:
“The aim of the present study was to examine 8 different popular nuts – pecan, pine, hazelnuts, pistachio, almonds, cashew, walnuts, and macadamia. Total content of phenolic compounds in nuts ranged from 5.9 (pistachio) to 432.9 (walnuts) mg/100 g.
Walnuts had the highest content of polymeric procyanidins, which are of great interest as important compounds in nutrition and biological activity, as they exhibit antioxidant, anti-inflammatory, antimicrobial, cardio- and neuroprotective action. Walnuts are good sources of fatty acids, especially omega-3 and omega-6.”
A second study compared the same eight tree nuts plus Brazil nuts and peanuts:
“The highest total content of all analyzed flavonoids was determined in walnuts (114.861 µg/g) with epicatechin the most abundant, while the lowest was in almonds (1.717 µg/g). Epicatechin has antioxidant, anti-inflammatory, antitumor, and anti-diabetic properties. Epicatechin has beneficial effects on the nervous system, enhances muscle performance, and improves cardiac function.”
Next, two systematic reviews and meta-analyses of human studies:
“We carried out a systematic review of cohort studies and randomized controlled trials (RCTs) investigating walnut consumption, compared with no or lower walnut consumption, including those with subjects from within the general population and those with existing health conditions, published from 2017 to 5 May 2021.
Evidence published since 2017 is consistent with previous research suggesting that walnut consumption improves lipid profiles and is associated with reduced CVD risk.
Evidence pointing to effects on blood pressure, inflammation, hemostatic markers, and glucose metabolism remains conflicting.
Evidence from human studies showing that walnut consumption may benefit cognitive health, which is needed to corroborate findings from animal studies, is now beginning to accumulate.”
“We aimed to perform a systematic review and meta-analysis of RCTs to thoroughly assess data concerning effects of walnut intake on selected markers of inflammation and metabolic syndrome in mature adults. Our findings showed that:
Walnut-enriched diets significantly decreased TG, TC, and LDL-C concentrations, while HDL-C levels were not significantly affected.
No significant changes were noticed on anthropometric, cardiometabolic, and glycemic indices after higher walnut consumption.
Inflammatory biomarkers did not record statistically significant results.”
https://www.mdpi.com/2076-3921/11/7/1412/htm “Walnut Intake Interventions Targeting Biomarkers of Metabolic Syndrome and Inflammation in Middle-Aged and Older Adults: A Systematic Review and Meta-Analysis of Randomized Controlled Trials”
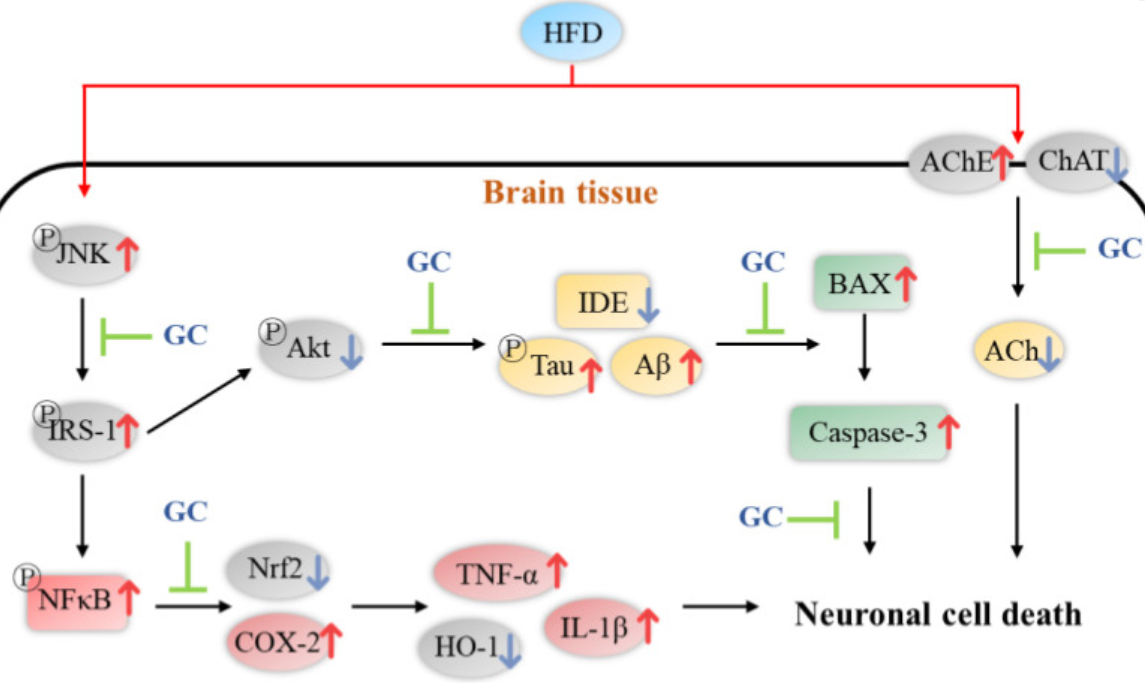
Finishing with a rodent study that gave subjects diabetes with a high-fat diet, then mixed two concentrations of walnut extract in with the treatment groups’ chow:
“This study was conducted to evaluate the protective effect of Gimcheon 1ho cultivar walnut (GC) on cerebral disorder by insulin resistance, oxidative stress, and inflammation in HFD-induced diabetic disorder mice. After HFD feed was supplied for 12 weeks, samples were orally ingested for 4 weeks to GC20 and GC50 groups (20 and 50 mg/kg of body weight, respectively).
Administration of GC improved mitochondrial membrane potential function, and suppressed oxidative stress in the brain.
GC inhibited hepatic and cerebral lipid peroxidation and the formation of serum AGEs, and increased serum antioxidant activity to improve HFD-induced oxidative stress.
The HFD group showed significant memory impairment in behavioral tests. On the other hand, administration of GC showed improvement in spatial learning and memory function.
Based on these physiological activities, GC showed protective effects against HFD-induced diabetic dysfunctions through complex and diverse pathways.”
https://www.mdpi.com/1420-3049/27/16/5316/htm “Walnut Prevents Cognitive Impairment by Regulating the Synaptic and Mitochondrial Dysfunction via JNK Signaling and Apoptosis Pathway in High-Fat Diet-Induced C57BL/6 Mice”
As mentioned in Week 127, I had biological age measured earlier this month, and received five reports two days ago on Sunday. Part of the company’s process is to follow up their reports (intrinsic aging, immune aging, pace of aging, telomere length, weight loss) with a consulting session to review and interpret, which lasted an hour yesterday.
Part of our conversation revolved around comparing my measurements with other customers. These people are a different population than people usually sampled for aging and other biomarkers, because people who pay to get their biological age measured probably actionably want to improve it.
We’ll see which items I asked the consultant to pass on to the company produce responses, and which interfere with their business or they’re too busy to get back to me. I offered more than a half-dozen specifics, but held back on items I didn’t think the consultant would adequately communicate.
I didn’t argue with the consultant’s recommendations for quercetin supplementation (at 4% bioavailability?) as part of a treatment for senescence (not measured in any of the reports?). I didn’t offer to follow-up with studies demonstrating yeast cell wall β-glucan (new to the consultant) effects on immune report findings here in my 19th year of taking it every day.
I did argue with their recommendation to take DHEA-S. I changed my mind about taking it a year and a half ago, but left blog posts up such as Take responsibility for your one precious life – DHEA for evidence that I’m learning.
Epigenetic clocks per The epigenetic clock theory of aging generally view biological aging as “an unintended consequence of both developmental programmes and maintenance programmes, the molecular footprints of which give rise to DNAm [DNA methylation] age estimators.”
So what would be appropriate anti-aging actions for customers to take? Should customers try to emulate youthful biological markers, and supplement DHEA-S to impact serum levels of insulin-like growth factor 1?
I don’t think so. Our bodies never evolved feedback mechanisms to determine “Time to stop the growth programs, you’ve survived to reproduction age.” Older people achieving teenagers’ DHEA-S levels and activating IGF-1 pathways, pretty much guarantees further biological aging as “an unintended consequence of both developmental programmes and maintenance programmes.”
It’s too early to recommend these biological aging measurements. We’ll see where it goes.
One good thing is the company wants their customers to tell them everything about what they’re doing. I exercise at least a half hour every day, eat Avena nuda oats for breakfast and AGE-less chicken vegetable soup for dinner, and take the following:
Before breakfast
– 3-day-old microwaved broccoli / red cabbage / mustard sprouts started from 10.7 grams of seeds, with nothing else an hour before or after
– Yeast cell wall β-glucan (Glucan 300), 1500 mg, with nothing else an hour before or after
Breakfast, lunch, and dinner
– Hyaluronic acid, Nature’s Lab, 1 serving total
– Boron, Swanson Triple Boron Complex, 9 mg total
Breakfast and dinner
– Acetyl-L-carnitine, 1 g total
– Balance oil, which blends linoleic acid 1400 mg with linolenic acid 350 mg, 2 times
– Betaine anhydrous, 3 g total
– Glucosamine hydroxychloride 1.5 g total, with chondroitin sulfate 1.2 g total
– Taurine, 2 g total
– 3-day-old Avena sativa oat sprouts started from 20 g seeds, 2 times
Breakfast only
– Minerals and vitamins, RDA mainly, Kirkland Signature Daily Multi
– D3 25 mcg
– Calcium alpha-ketoglutarate 1 g
Lunch only
– Vitamin K2 MK-7 600 mcg
Dinner only
– D3 50 mcg
– Zinc monomethionine 30 mg with 0.3 mg copper
– Lutein 25 mg with 5 mg zeaxanthin
“We use data from a safety study (n = 18, mean age 74) to investigate whether human umbilical cord plasma concentrate (hereinafter Plasma Concentrate) injected weekly (1 ml intramuscular) into elderly human subjects over a 10-week period affects different biomarkers, including epigenetic age measures, standard clinical biomarkers of organ dysfunction, mitochondrial DNA copy number (mtDNA-CN), and leukocyte telomere length.
More than 20 clinical biomarkers were significantly and beneficially altered. Telomere length and mtDNA-CN were not significantly affected by treatment.
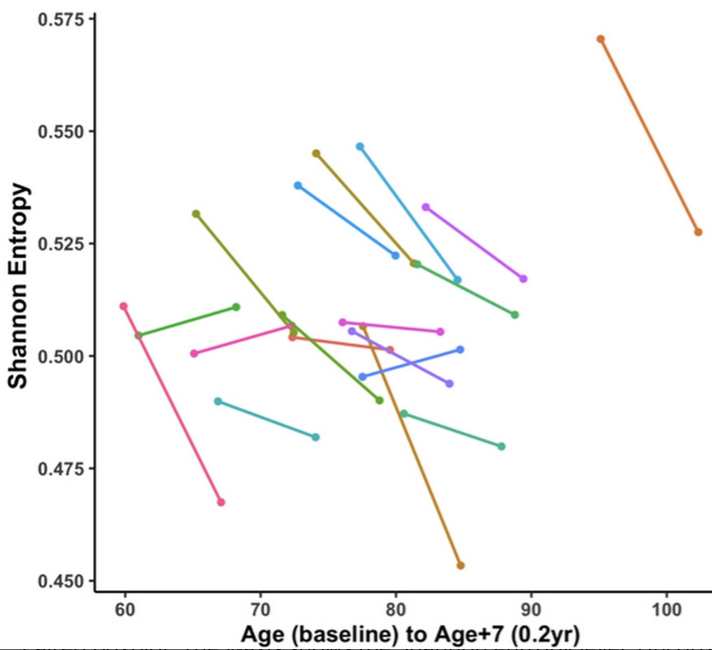
An increase in entropy means that the methylome becomes noisier. We found that entropy was significantly decreased after treatment. Decreased entropy may implicate rejuvenation of the epigenetic landscape after plasma concentrate treatments.
Treatment reduced DNA methylation-based GrimAge by an average of 0.82 years, suggesting a reduction in morbidity and mortality risk. By contrast, no significant results could be observed for epigenetic clocks that estimate chronological age.
Our study lends credence to the notion that there are youth-promoting factors in the secretome of umbilical cord plasma. This conclusion has also been reached by other researchers that have provided treatment with stem cells, which do not work by plasma dilution but primarily by providing humoral factors and changing the microenvironment of cells and tissues. While there may be youth-promoting microvesicles or humoral factors that are at work, we do not want to rule out the possibility that it is ‘young and undamaged’ albumin that leads to the improvements noted, especially in light of recent evidence for such a mechanism.
This first human epigenetic clock study of plasma concentrate treatments revealed age-reversal effects according to a well-established DNA methylation-based estimator of morbidity and mortality risk. Future placebo-controlled replication studies are warranted with a larger number of participants over a longer study period, which our laboratory has undertaken to pursue.”