People will forgive you for being wrong, but they will never forgive you for being right – especially if events prove you right while proving them wrong. Thomas Sowell
A 2026 study investigated aging through the use of transcriptomics:
“By constructing transcriptomic clocks of expected mortality across >11,000 samples from mouse, rat, macaque and human, we provide a unified framework that integrates age-associated transcriptional change with the direction and magnitude of lifespan modulation by interventions, enabling a quantitative and biologically interpretable readout of health status.
While chronological clocks captured many detrimental conditions (e.g., chronic diseases or certain short-lived genetic models), they were comparatively less responsive to lifespan-extending interventions, consistent with reports that longevity treatments often only modestly reverse age-associated transcriptomic signatures. In contrast, mortality clocks robustly distinguished both short- and long-lived models and showed strong associations with human time-to-death, approaching the predictive performance of second-generation DNAm mortality clocks while remaining biologically interpretable.
Our results support and extend DNAm-based observations that heterochronic parabiosis in old animals and early embryogenesis can induce molecular age deceleration. In old heterochronic parabionts, mortality clocks detected a sustained tAge reduction that persisted for 2 months after detachment from young partners. Together, these findings support the view that early development contains a conserved rejuvenation-like phase, and that systemic environment can partially reset molecular age later in life.
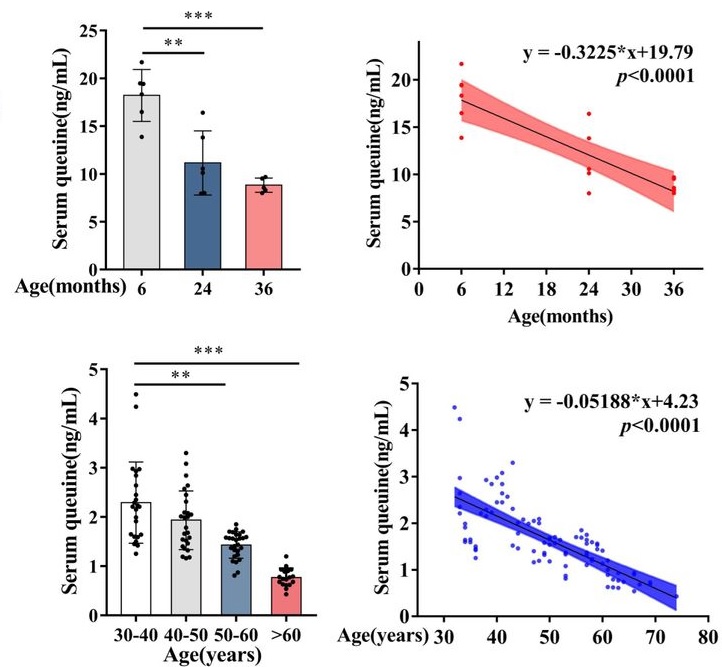
“The contribution of transfer RNA (tRNA)-specific modifications to aging remains largely unexplored. We systematically profile tRNA modifications across multiple organs, species, and senescence models, and identify mannosyl-queuosine (manQ) as the first tRNA-specific modification that consistently declines with age.
Across species, queuine supplementation extends lifespan and enhances healthspan. In naturally aging mice, long-term oral administration beginning at 16-months-old (human equivalent 50 years) extends mean lifespan by 15.3%, reduces DNA methylation age, improves cognitive and motor performance, strengthens antioxidant defenses, remodels the gut microbiota, and alleviates inflammation and metabolic dysfunction without detectable toxicity.
These findings establish tRNA epitranscriptomic remodeling as a previously unrecognized layer of aging regulation, and identify restoration of manQ through queuine supplementation as a multi-system strategy to delay aging.
manQ hypomodification is selective rather than reflecting global tRNA depletion. Aging preferentially reduces the manQ-containing tRNAAsp fragment while leaving the corresponding unmodified tRNAAsp fragment, and other queuosine-modified tRNAs, relatively unchanged.
This pattern supports a regulated defect in modification homeostasis rather than a generalized change in transcript abundance. Such specificity argues that manQ loss is not merely a passive consequence of tissue degeneration, but instead represents a conserved, biologically meaningful aging-associated event with mechanistic impact.
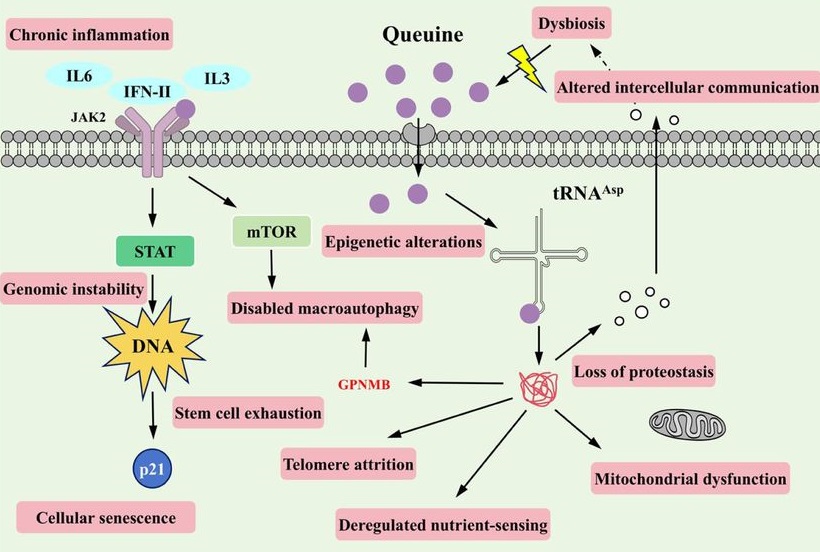
Because proteostasis intersects with multiple canonical hallmarks (e.g. mitochondrial dysfunction, impaired stress resilience, and altered intercellular communication), translation-coupled proteome destabilization offers a unifying explanation for how a single tRNA modification defect can elicit multi-system consequences. In this view, manQ decline is not merely one of many molecular changes observed in aging, but rather a proximate determinant capable of amplifying downstream hallmarks through a common axis of proteome quality control.
Our findings further suggest that manQ depletion may engage self-reinforcing feedback loops that accelerate aging trajectories. This architecture offers a conceptual framework in which aging progressively erodes ‘epitranscriptomic integrity’ at the tRNA level, pushing translation toward an error-prone regime that accelerates proteostatic collapse and functional decline.
A distinctive implication of this work is that queuine introduces a microbiota-host epitranscriptomic axis into aging biology. Queuine is produced by gut microbiota and cannot be synthesized de novo by mammals. These findings expand the conceptual scope of geroscience by placing a microbiota-derived nutrient upstream of translational quality control.
Queuine supplementation offers a distinct therapeutic logic: rather than modulating a single signaling cascade, it restores a tRNA modification state that governs translational fidelity – an upstream determinant of proteome quality that can, in principle, influence multiple downstream hallmarks concurrently. These findings highlight an intervention paradigm centered on restoring molecular fidelity, rather than suppressing a single downstream phenotype, as a strategy to delay systemic aging.”
A 2026 rodent study investigated sulforaphane’s ability to affect ALS-like symptoms:
“The objective of this study was to evaluate neuroprotective efficacy and safety of sulforaphane (SUFP) in a methylmercury (MMHg⁺)-induced preclinical rat model of amyotrophic lateral sclerosis (ALS). ALS is characterized by progressive motor neuron degeneration and muscle wasting, leading to impairments in gait, swallowing, salivation, and routine motor activities.
64 animals were classified into eight groups: 1st: normal control, 2nd: vehicle control; 3rd: SUFP perse (4 mg/kg, i.p.), 4th: MMHg + (5 mg/kg, p.o.), 5th: MMHg + 5 + SUFP (2 mg/kg, i.p.), 6th: MMHg+ 5 + SUFP (4 mg/kg, i.p.), 7th: MMHg+ 5 + omaveloxolone (OVX) (30 mg/kg, i.p.), and 8th: MMHg + 5 + dimethyl fumarate (DIMT) (50 mg/kg, i.p.). Neurotoxin MMHg + was orally administered at 5 mg/kg for the first 21 days. For the next 22 days, SUFP, OVX, and DIMT were administered intraperitoneally (i.p.).
SUFP modulates neurotransmitter levels such as acetylcholine (A), dopamine (B), GABA (C), glutamate (D), and serotonin (E).
SUFP4 exerted broad neuroprotective effects in ALS pathology by restoring antioxidant proteins (Nrf2, HO-1, SIRT1), suppressing apoptotic (Bax, caspase-3, Bcl-2) and inflammatory markers (TNF-α, IL-1β), and enhancing the anti-inflammatory cytokine IL-10. It also downregulated stress-related signaling pathways (PI3K/Akt, p75NTRECD, MAPKs) associated with neurodegeneration. These molecular effects translated into meaningful functional recovery, as evidenced by improvements in grip strength, locomotor performance, spatial memory, and depressive-like behavior.
Histopathological evaluation demonstrated attenuation of demyelination and preservation of neuronal architecture including the cerebral cortex, hippocampus, striatum, midbrain, and cerebellum. Beyond central neuroprotection, SUFP exerted systemic benefits by normalizing hepatic enzymes, improving skeletal muscle integrity, restoring redox balance, stabilizing neurofilament and myelin-associated proteins, and correcting hematological alterations.
Despite limitations related to study duration and animal sex, this work strongly positions SUFP as a promising, multi-target therapeutic candidate for ALS with both neural and systemic protective efficacy.”
https://link.springer.com/article/10.1007/s12035-026-05683-5 “Sulforaphane-Mediated Multitarget Therapeutic Effects in Methylmercury-Induced ALS-Like Pathology: Comparative Analysis and Multifaceted Approach to Neuroprotection and Systemic Recovery” (not freely available) Thanks to Dr. Sidharth Mehan for providing a copy.
Unlike A Nrf2 treatment for ALS?, this study didn’t present evidence that its treatment compound was effective for preventing ALS. For one thing, currently-known disease factors involving heat shock proteins and associated genes, some of which are Nrf2 targets, weren’t investigated.
Two Nrf2 activators were used in both studies as comparators of Nrf2 activation effects. Neither omaveloxolone nor dimethyl fumarate are ALS causal treatments, though, and have undesirable side effects.
A human equivalent of this study’s higher sulforaphane dose is ((4 mg x .162) x 70 kg) = 45 mg. 45 mg of sulforaphane might be too much to consistently take at one time because of unpalatability. But I documented taking an estimated 52 mg for a year during 2020-2021 by eating microwaved 3-day-old broccoli sprouts twice a day.
A 2026 review subject was mechanisms and therapeutic potential for Nrf2 activators in combination with mesenchymal stem cells:
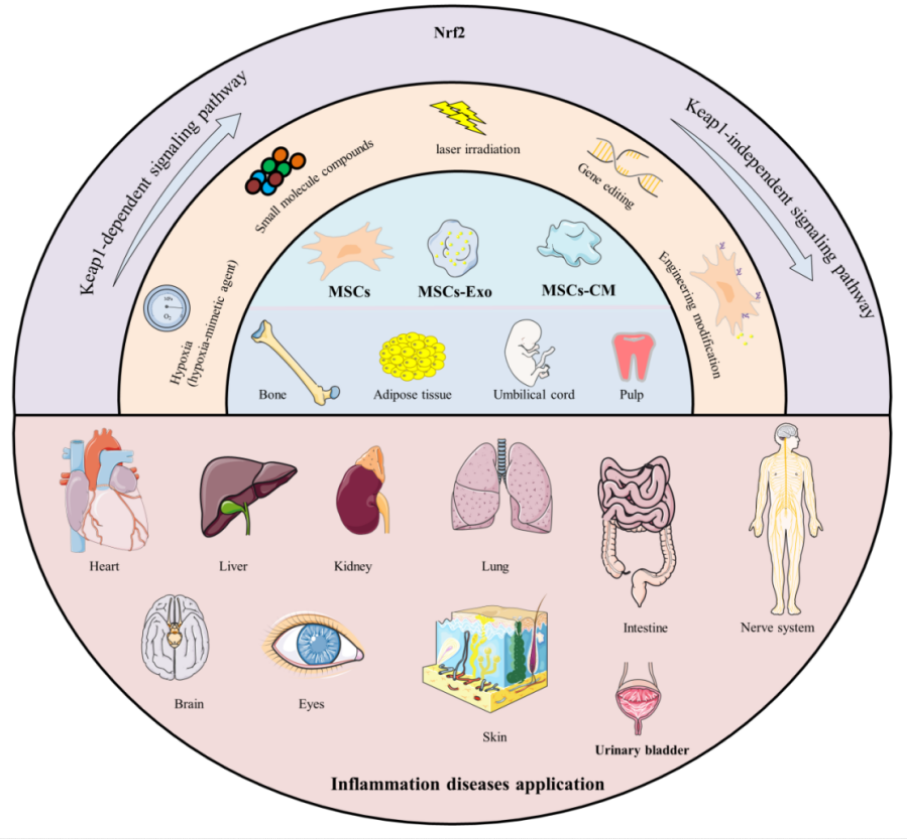
“Mesenchymal stromal/stem cells (MSCs) are multipotent stem cells that can be isolated from various tissues – such as bone marrow (BM), umbilical cord (UC), adipose tissue (AD), dental pulp (DP), hair follicle (HF), and placenta – and differentiated into multiple lineages under appropriate conditions. Their functional repertoire includes immunomodulation, homing, and differentiation, which collectively help establish a balanced inflammatory and regenerative niche within damaged tissues during severe inflammation. MSCs-derived extracellular vesicles (MSCs-EVs) and conditioned medium (MSCs-CM) play remarkable roles, exhibiting potent anti-inflammatory and antioxidant properties that offer novel therapeutic alternatives for inflammatory diseases.
Therapeutic capacity of MSCs in inflammatory conditions is increasingly attributed to their potent paracrine activity rather than solely to their differentiation potential. A key mechanism underlying this paracrine effect is activation of the Nrf2 antioxidant pathway.
MSCs and their secreted products including exosomes (Exos), EVs, and CM, activate Nrf2 through multi-dimensional/target mechanisms, thereby enhancing cellular antioxidant defenses, modulating immune responses, and promoting tissue repair. It is noteworthy that therapeutic efficacy of MSCs and their derivatives can be enhanced through external modulation, including pretreatment with natural compounds.
Preconditioning refers to brief treatment of MSCs or their derivatives with physical, chemical, or biological factors prior to application, aiming to enhance their ability to counteract oxidative stress and improve their therapeutic efficacy. Flavonoids precondition and prime MSCs via the direct Keap1-Nrf2 pathway or indirect PI3K-Akt pathway, which enhances cellular resilience to adverse conditions by reducing apoptosis and promoting survival. Primed MSCs, in turn, remodel the microenvironment through an altered secretory profile, releasing bioactive factors that create more favorable conditions for their own persistence.
The core logic of these strategies lies in simulating or inducing adaptive stress, such as employing specific chemical molecules or drug stimuli, or utilizing physical / microenvironmental preconditioning to mimic specific physical conditions of the in vivo injury environment. The most straightforward strategy is overexpression of Nrf2 or its key downstream effector molecules.
The majority of existing studies remain at the level of observing correlations with Nrf2 upregulation, and there is still a lack of precise causal validation regarding key upstream signals – such as specific cytokines, miRNAs, or proteins – through which MSCs or derivatives initiate Nrf2 activation. Mechanistic insights are predominantly derived from in vivo or rodent (mouse/rat) model experiments, with a notable absence of clinical validation, insufficient long-term safety and pharmacokinetic data, and a lack of standardization in administration routes and dosages, all of which hinder clinical translation.
The essential role of the Nrf2 pathway has not been rigorously confirmed, as most studies have not employed reverse genetic validation using Nrf2-knockout animals or specific inhibitors. Consequently, it remains unclear whether therapeutic effects are necessarily and exclusively dependent on Nrf2, and potential synergistic contributions from other pathways may have been overlooked.
Most natural flavonoids face challenges such as low oral bioavailability, rapid metabolism, and poor targeting. Numerous challenges remain to be addressed in order to translate these promising preclinical findings into clinical practice. Future research should focus on the following aspects:
Elucidating precise upstream molecular mechanisms by which MSCs activate Nrf2;
Employing more clinically relevant chronic disorder models;
Systematically evaluating long-term safety, optimal delivery strategies (including dosage and route of administration), and immunogenicity of MSCs-based therapies;
Validating selection criteria (optimal source), quality control, batch-to-batch consistency of MSCs, and addressing regulatory and ethical barriers to clinical translation; and
Integrating molecular docking, ADMET (Absorption, Distribution, Metabolism, Excretion, Toxicity) prediction, and in vitro and in vivo validation to further elucidate regulatory effects of flavonoids and enhance understanding of their mechanisms of action.”
This paper was overly long at 127 pages, so I focused on the later sections. None of these treatments are currently ready for clinical trials.
I also didn’t mention specific flavonoids as Nrf2 activators. It’s beyond a reviewer’s task to rank Nrf2 activators, and a study’s researchers seldom address why they used a poorly-activating flavonoid instead of a higher-ranked natural plant compound such as sulforaphane.
A 2026 paper provided details of a 2020-2023 human trial of broccoli sprouts:
“In a 42-month randomized, double-blind, placebo-controlled trial, 26 participants aged 63–90 years with memory impairment were randomly assigned to receive either 30 mg/day of glucoraphanin (GLR) or placebo. The primary outcome was the change in Memory Performance Index (MPI) scores from the mild cognitive impairment (MCI) screen. This study evaluated the long-term efficacy of GLR supplementation on cognitive function in older adults at an elevated risk for Alzheimer’s disease (AD), including those with MCI.
Participants were instructed to take three capsules of either the GLR or placebo supplements daily for 42 months. The GLR supplement contained 30 mg of GLR purified from broccoli sprouts, along with 120 mg of mustard powder per three capsules. Mustard powder was included as a source of exogenous active myrosinase to enhance the enzymatic conversion of GLR to sulforaphane. The placebo supplement contained 0 mg of GLR.
No significant group difference was observed in the initial 6 months. A marginal difference in favor of GLR appeared in the later phase (30 and 42 months), including the 42-month endpoint.
The GLR group demonstrated superior performance on immediate recall and delayed free recall tests. MCI participants showed a greater MPI improvement with GLR.
Long-term GLR supplementation may help preserve cognitive function in individuals at elevated risk for AD, particularly those with MCI. Larger trials are warranted to confirm efficacy and clarify underlying mechanisms.”
This study was funded by the supplement manufacturer. There was no explanation of what the supplement’s “purified from broccoli sprouts” entails. Also, I didn’t mention results of voluntary group exercise because there was a long gap in the participants’ data due to government response to covid.
For comparison of this study’s 30 mg glucoraphanin dose, Our model clinical trial for Changing to a youthful phenotype with broccoli sprouts provided 30 grams of fresh broccoli sprouts that contained an estimated 51 mg of glucoraphanin for ten weeks. That study’s corresponding coauthor said of their 30 gram broccoli sprouts dose in Understanding a clinical trial’s broccoli sprout amount that “When we carried out tests with consumers, previous to the bioavailability studies, higher amounts per day were not easy to consume and to get eaten by participants.” There was no rationale provided for this study’s 30 mg dose other than citing two previous human studies that also used a 30 mg glucoraphanin dose.
For comparison of this study’s 120 mg mustard seed powder dose, my daily cruciferous food intake since five and a half years ago includes sprouted yellow mustard seeds started with 3.5 grams of seeds, along with sprouted broccoli and red cabbage started with 3.6 grams of each vegetable’s seeds, all sprouted for three days. I haven’t seen studies that show sprouting has effects on myrosinase enzyme activity.
This study cited Does sulforaphane reach the colon? which used 2% mustard seed powder to create sulforaphane from glucoraphanin. This study’s 120 mg mustard seed powder / 30 mg glucoraphanin is a lot more than 2%. My daily sprout intake started from 3.5 g mustard seeds / (3.6 g broccoli seeds +3.6 g red cabbage seeds) is also a lot more than 2%.
I’ve changed some items along the way, switching supplier from True Leaf to Johnny’s for organic broccoli seeds, and from non-organic Red Acre red cabbage seeds to True Leaf organic red cabbage seeds. I recently had to find another supplier of organic yellow mustard seeds when Naturevibe stopped carrying that product. I tried Food to Live, but their yellow mustard seeds when sprouted aren’t mild. I’ll next try Frontier Co-op to see if those are mild as advertised.
Starting this blog’s twelfth year by curating a poorly-done 2026 review of Nrf2 and its capability to change a person’s development of Parkinson’s disease. I’ll emphasize precedent conditions that if not effectively dealt with in youth, can’t prevent PD from occurring at some later life stage.
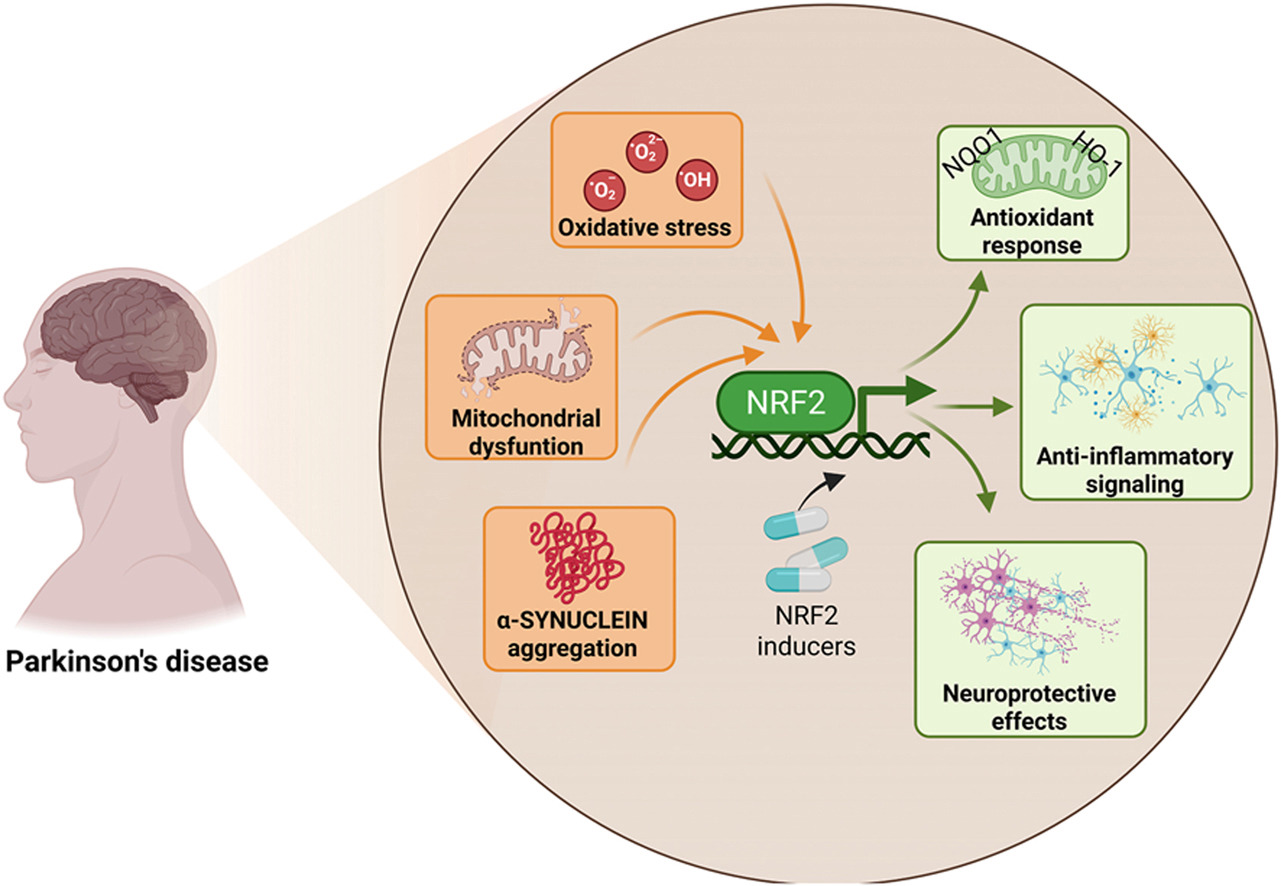
“This review explicitly examines how age-associated decline in NRF2 responsiveness intersects with redox imbalance, mitochondrial dysfunction, proteostatic failure, and neuroinflammation, core mechanisms shared between aging and PD. PD unfolds through a complex interplay of cellular stress and immune responses. Oxidative stress, mitochondrial dysfunction, and chronic neuroinflammation converge to damage dopaminergic neurons, with microglia playing a central role in amplifying this injury.
NRF2 emerges as a key regulator of antioxidant defenses, inflammatory balance, and mitochondrial protection, offering a promising target for clinical intervention. NRF2 activity favors the anti-inflammatory microglial over the pro-inflammatory phenotype. Decline in NRF2 inducibility with age impairs microglial clearance, promotes neuroinflammation, and reduces antioxidant defenses, while NRF2 activation restores protective functions and offers a promising therapeutic target.
Strategies aimed at restoring or enhancing NRF2 activity hold significant promise as disease-modifying interventions, not only to slow PD progression but also to promote resilience against the broader spectrum of age-associated neurodegenerative and inflammatory conditions.”
This review only gave lip service to PD progression outside of the brain, as if the importance of prodromal factors to a person’s neurodegeneration such as dysfunction in gut, eyes, skin, and olfactory systems can be minimized. But failure to recognize early what will doom a person to be unable to recover health in later decades is disingenuous. Since these reviewers omitted early interventions into PD prodromal factors, the best they came up with was interventions to “slow PD progression.”
Maybe these reviewers felt it would be outside the scope of this review to discuss early non-brain PD factors for more than one sentence? However, while PD is defined by striatal brain neurons, Nrf2 activity is much less in brain and central nervous system neurons than elsewhere in the body per Nrf2 Week #2: Neurons.
I disagree with these reviewers’ self-imposed emphasis on aging. Repeating ‘age-associated’ numerous times seemed as if they wanted to influence the reader into thinking age in and of itself was a cause for PD, rather than an imputed mathematical correlation. Their emphasis led to dumb mentions such as senolytics for no apparent reason than senescence is a ‘hallmark of aging’, and to meaningless ‘diseasome of aging’ characterizations, and to ignoring the existence of early non-age-associated PD diagnoses in 20- and 30-year-olds.
Whatever it takes to get published, I’d guess. Or maybe it’s that the number of omissions and useless points a review paper makes increases with the number of reviewers and their sponsors’ agendas.
For example, why was it permissible to dedicate lip service to ‘exposome’ factors like microplastics, environmental pollution, and viruses, but it’s still not permitted in 2026 to discuss research into the impacts on vascular disease and neurodegeneration of lipid nanoparticles and DNA contamination in what a large number of humans were exposed to by injected pharmaceuticals starting in late 2020? Not to mention two studies published in 2024 of over 2.5 million people whose incidences of neurologic issues, mild cognitive impairment, and Alzheimer’s disease rapidly increased after ‘vaccination’?
I’ve mentioned in this blog many times how it’s every human’s choice whether or not we take responsibility for our own one precious life. I suggest, if it’s not too late, do that for your children’s lives, too.
Here are five 2025 human ergothioneine studies, starting with a clinical trial of healthy older adults:
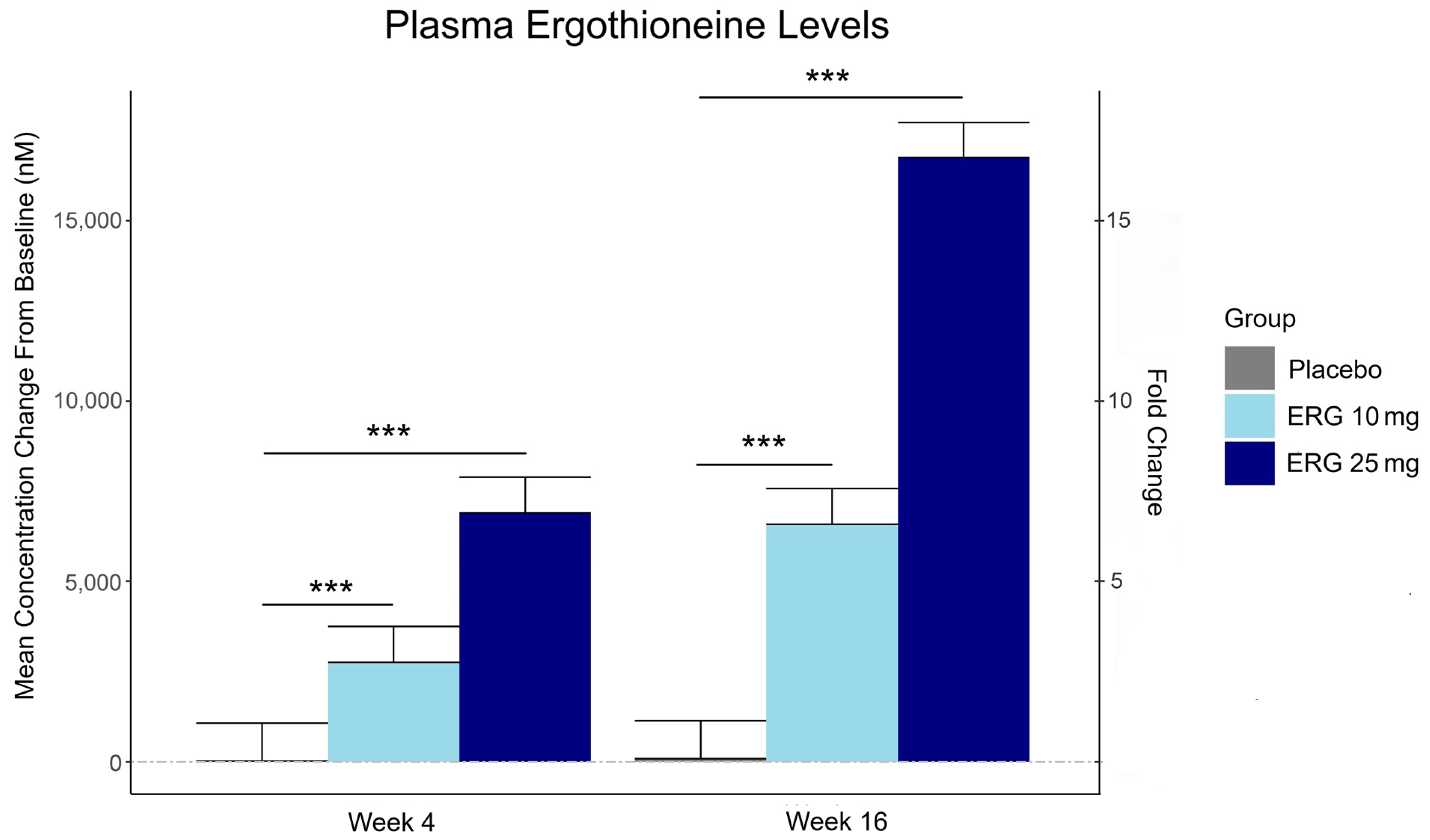
“In this 16-week randomized, double-blind, placebo-controlled trial, 147 adults aged 55–79 with subjective memory complaints received ergothioneine (10 mg or 25 mg/day ErgoActive®) or placebo. Across all the groups, approximately 73% of participants in each group were female, with a median age of 69 years.
The primary outcome was the change in composite memory. Secondary outcomes included other cognitive domains, subjective memory and sleep quality, and blood biomarkers. At baseline, participants showed slightly above-average cognitive function (neurocognitive index median = 105), with plasma ergothioneine levels of median = 1154 nM.
Although not synthesized in the human body, ergothioneine is efficiently absorbed via the OCTN1 transporter (also known as the ergothioneine transporter, or ETT), which is expressed in many tissues, including the intestine, red blood cells, kidneys, bone marrow, immune cells, skin, and brain. This transporter enables ergothioneine to accumulate in high concentrations in organs vulnerable to oxidative stress and inflammation. Ergothioneine has multiple cellular protective functions, including scavenging reactive oxygen species, chelating redox-active metals, suppressing pro-inflammatory signaling, and protecting mitochondrial function.
Plasma ergothioneine increased by ~3- and ~6-fold for 10 mg, and ~6- and ~16-fold for 25 mg, at weeks 4 and 16, respectively.
While the primary outcome, composite memory, showed early improvement in the 25 mg group compared to baseline, this effect was not sustained and did not differ from placebo. Reaction time showed a significant treatment-by-time interaction favoring ergothioneine, yet the between-group differences were not significant, suggesting that any potential benefits were modest and require validation in larger or longer studies.
Other cognitive effects observed were primarily within-group and not consistently dose-responsive, highlighting the challenge of detecting objective cognitive changes over a relatively short study duration in high-functioning healthy populations. However, positive effects of ergothioneine supplementation were observed on subjective measures of prospective memory and sleep initiation that were not seen in the placebo group.
This trial adds to the growing body of evidence supporting the favorable safety profile of ergothioneine. No adverse events attributable to ergothioneine were reported. Additionally, we observed potential hepatoprotective effects, with significant reductions in the plasma AST and ALT levels, particularly among males in the ERG 25 mg group.”
https://www.mdpi.com/1661-3821/5/3/15 “The Effect of Ergothioneine Supplementation on Cognitive Function, Memory, and Sleep in Older Adults with Subjective Memory Complaints: A Randomized Placebo-Controlled Trial”
The third graphic for Ergothioneine dosing, Part 2 showed a human study where a 25 mg dosing stopped after Day 7, but the plasma ergothioneine level stayed significantly higher than baseline through Day 35.
The second graphic for Ergothioneine dosing, Part 2 was a male mouse experiment where plasma ergothioneine levels of a human equivalent 22 mg to 28 mg daily dose kept rising through 92 weeks.
This trial couldn’t explain the desirability of a 25 mg daily dose that was likely (per the second and third graphics for Ergothioneine dosing, Part 2) to sustain the subjects’ increased plasma ergothioneine levels well after the trial ended at Week 16. What effects can be expected from a sustained plasma ergothioneine level that’s 16 times higher than the subjects’ initial levels? Were these 16-fold sustained plasma ergothioneine levels better or worse than the 6-fold increases in the 10 mg group, both of which were likely to continue past the trial’s end?
A representative of the trial’s sponsoring company talked a little more about the trial in this interview:
Another clinical trial investigated ergothioneine’s effects on skin:
“We conducted an 8-week, randomized, double-blind, placebo-controlled clinical trial to evaluate effects of daily oral supplementation with 30 mg of ergothioneine (DR.ERGO®) on skin parameters in healthy adult women aged 35–59 years who reported subjective signs of skin aging. Objective measurements including melanin and erythema indices, skin glossiness, elasticity, and wrinkle and pigmentation counts were used to comprehensively evaluate changes in skin condition.
The OCTN1 transporter is preferentially expressed in basal and granular epidermal layers, where cellular renewal and barrier maintenance are most active. Once internalized, ergothioneine localizes to mitochondria, where it directly scavenges reactive oxygen species (ROS) and protects mitochondrial DNA from UV- and inflammation-induced damage.
At the signaling level, ergothioneine activates key protective pathways such as the Nrf2/ARE axis, enhancing expression of antioxidant enzymes including HO-1, NQO1, and γ-GCLC. These enzymes contribute to redox homeostasis and glutathione regeneration, reinforcing cellular defense systems against photoaging and environmental insult.
In parallel, ergothioneine modulates the PI3K/Akt/Nrf2 and SIRT1/Nrf2 pathways, which are implicated in collagen preservation, inflammation resolution, and mitochondrial maintenance. These pathways converge to reduce matrix metalloproteinase (MMP) activity, enhance collagen synthesis, and suppress pro-inflammatory cytokines (TNF-α, IL-6, IL-1β), all of which are central to maintaining skin structure and function.
Compared to placebo, the DR.ERGO® ergothioneine group showed significantly greater improvements in melanin and erythema reduction, skin glossiness, elasticity, and wrinkle and spot reduction. No adverse events were reported.
These findings corroborate and extend previous clinical evidence from (Hanayama et al., 2024), who investigated an ergothioneine-rich mushroom extract (Pleurotus sp., 25 mg ergothioneine/day) in a 12-week randomized double-blind trial, and (Chunyue Zhang, 2023), who examined pure ergothioneine supplementation (25 mg/day) in a 4-week open-label study. We contextualized our results within this existing literature by comparing key outcomes.
Several limitations should be acknowledged:
The study cohort consisted solely of Japanese women aged 35–59 years, which may limit generalizability across sexes, ethnicities, and age groups.
The 8-week intervention period, while sufficient to detect short-term effects, does not allow conclusions about the sustainability of benefits or the risk of relapse upon discontinuation.
The placebo group also showed modest improvements in self-perception, highlighting the well-documented placebo response in beauty and wellness studies.
This study focused on a single daily dosage (30 mg/day) without evaluating dose–response relationships, and hydration-specific endpoints such as corneometry or transepidermal water loss (TEWL) were not included.”
Two clinical trials investigated ergothioneine’s effects on sleep quality:
“A four-week administration of 20 mg/day ergothioneine (EGT), a strong antioxidant, improves sleep quality; however, its effect at lower doses remains unclear. This study estimated the lower effective doses of EGT using a physiologically based pharmacokinetic (PBPK) model in two clinical trials.
In Study 1, participants received 5 or 10 mg/day of EGT for 8 weeks, and their plasma and blood EGT concentrations were measured. An optimized PBPK model incorporating absorption, distribution, and excretion was assembled. Our results showed that 8 mg/day of EGT for 16 weeks was optimal for attaining an effective plasma EGT concentration.
In Study 2, a randomized, double-blind, placebo-controlled study, participants received 8 mg/day EGT or a placebo for 16 weeks. The subjective sleep quality was significantly improved in the EGT group than in the placebo group.
In mammals, EGT is not generated in the body but is acquired from the diet via the carnitine/organic cation transporter OCTN1/SLC22A4. Its plasma concentration after oral administration is quite stable and gradually increases after repeated dosing on a multi-day basis.
Blood concentrations of EGT increase after Day 8 when EGT intake is interrupted, and they continue to increase until Day 35. The delayed increase in EGT concentration in the blood, compared with that in the plasma, can be interpreted as its efficient uptake by undifferentiated blood cells, which express high levels of OCTN1/SLC22A4 in the bone marrow, and subsequent differentiation to mature blood cells that enter the circulation. This may imply the nonlinear absorption, distribution, and excretion of EGT owing to saturation of the transporter at higher concentrations, potentially leading to difficulty in model construction.
This is the first study to propose a strategy to estimate lower effective doses based on the PBPK model.”
The bolded section above referenced a 2016 study / third graphic for Ergothioneine dosing, Part 2, where a 25 mg dosing stopped after Day 7, but the plasma ergothioneine level stayed high through Day 35. I didn’t see that the referenced 2004 and 2010 studies addressed this 2016 finding.
I also didn’t see that this study’s mathematical model accounted for saturation of the OCTN1 transporter or other effects, such as a very small ergothioneine clearance rate. Okay, lower the ergothioneine dose, and achieve a lower persistent plasma ergothioneine level, to what benefit?
“The present study demonstrated that OCTN1 is associated with myeloid cells rather than lymphoid cells, and especially with erythroid-lineage cells at the transition stage from immature erythroid cells to peripheral mature erythrocytes.”
Persistent high ergothioneine levels aren’t costless. Skewing bone marrow stem cells and progenitor cells toward a myeloid lineage is done at the expense of lymphocytes, T cells, B cells, and other lymphoid lineages.
Where are the studies that examine these tradeoffs? Subjective sleep quality in this study and sleep initiation in the first study above aren’t sufficiently explanatory.
A study investigated associations of plasma ergothioneine levels and cognitive changes in older adults over a two-year period:
“Observational studies have found that lower plasma levels of ergothioneine (ET) are significantly associated with higher risks of neurodegenerative diseases. However, several knowledge gaps remain:
Most of the above studies were based on cross-sectional study design, and potential reverse causation cannot be excluded. It has been suggested that plasma ET declines concomitantly with the deterioration of cognitive function.
Since the impact of a single dietary factor on health is mild, it is prone to be affected by the baseline characteristics of subjects (such as sex, educational level, disease status and gene polymorphism). However, no study has systematically evaluated potential effect modifiers on the association between ET levels and cognitive function.
The dose-response distribution between ET and cognitive function remains undetermined.
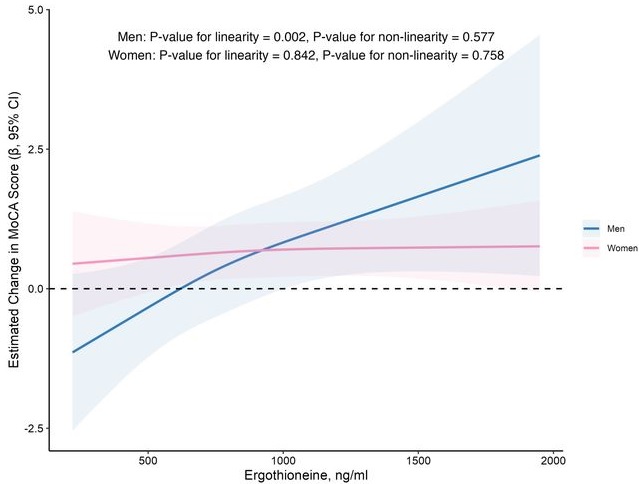
In this prospective cohort study of 1,131 community-dwelling older adults (mean age 69 years), higher baseline plasma ET levels were significantly associated with slower cognitive decline, as assessed by Montreal Cognitive Assessment (MoCA) scores, during a 2-year follow-up period.
When the plasma concentration of ET exceeds 1,000 ng/mL, the decline in cognitive function significantly slows down. However, this association has only been observed in men.
Domain-specific analysis found that the observed ET-MoCA association was mainly driven by the temporary slowdown in the decline of visuospatial/executive and delayed recall. Impaired delayed recall represents one of the earliest and most sensitive cognitive markers of dementia progression, predictive of conversion from MCI to dementia. The preferential preservation of this function by ET suggests targeted neuroprotective effects within the hippocampus.
Visual inspection of the spline curves revealed a potential plateauing effect at ET concentrations ≥1,000 ng/mL in the total population.
Baseline ET concentrations differed between men and women. Most men (81.5%) had concentrations below 1,000 ng/mL (median 754.2, IQR 592.0–937.9 ng/mL). Women exhibited substantially higher median plasma ET concentrations than men, with 35.7% of women exceeded 1,000 ng/mL (median 890.1, IQR 709.7–1,095.6 ng/mL).
Our study included only participants with normal cognitive function, and the results remained robust even after excluding those with baseline cognitive function at the lower end of the normal range. Collectively, our findings support that low ET intake occurs prior to cognitive decline.
Our findings indicate that higher plasma ET levels are significantly associated with slower cognitive decline independent of confounders in non-demented community-dwelling elderly participants, with such association observed in men but not women. Dose-response curves indicated plateauing effects above 1000 ng/mL.”
The average age of this study and the first trial above were both 69 years. Since the first trial’s participants showed slightly above-average cognitive function (neurocognitive index median = 105), with plasma ergothioneine levels of median = 1154 nM at baseline, and this study showed plateauing effects above 1000 ng/mL, I wonder how raising plasma ergothioneine levels above 1000 ng/mL could possibly show a net benefit for older people? What are the trade-offs for older people between potentially increasing slightly above-average cognitive function with ergothioneine and its other effects from saturating their OCTN1 transporter?
This study is at its preprint stage. I’m interested to see if its peer review prompts these researchers to also investigate the common finding that people who are most deficient at baseline have the greatest improvements. If so, would these sex-specific associations still hold?
Wrapping up with a study that investigated associations of serum ergothioneine levels with the risk of developing dementia:
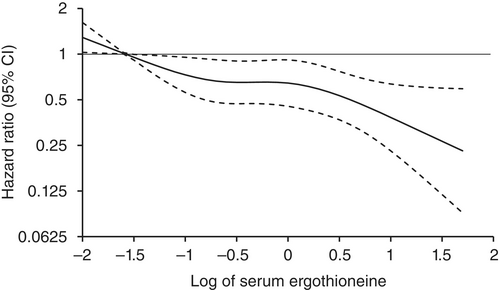
“1344 Japanese community-residents aged 65 years and over, comprising 765 women and 579 men, without dementia at baseline were followed prospectively for a median of 11.2 years.
During follow-up, 273 participants developed all-cause dementia. Among them, 201 had Alzheimer’s disease (AD) and 72 had non-Alzheimer’s disease (non-AD) dementia.
Age- and sex-adjusted hazard ratios (HRs) for all-cause dementia, AD, and non-AD dementia decreased progressively across increasing quartiles of serum ergothioneine. These associations remained significant after adjustment for a wide range of cardiovascular, lifestyle, and dietary factors, including daily vegetable intake.
In subgroup analysis, association between serum ergothioneine levels and the risk of dementia tended to be weaker in older participants and in women:
In older individuals, cumulative burden of multiple risk factors such as hypertension, diabetes mellitus, and smoking may contribute to both neurodegenerative and vascular pathology, potentially diminishing the relative influence of ergothioneine.
In women, postmenopausal hormonal changes, particularly the decline in estrogen, have been associated with increased oxidative stress and a higher vulnerability to neurodegenerative changes.
Several limitations should be noted:
Since serum ergothioneine levels and other risk factors were measured only at baseline, we could not evaluate the changes of serum ergothioneine levels during the follow-up period. Lifestyle modifications during follow-up could have influenced serum ergothioneine levels and other risk factors. In addition, serum ergothioneine level was measured only once, and from a sample.
We cannot rule out residual confounding factors, such as other nutrients in mushrooms and socioeconomic status.
There is a possibility that dementia cases at the prodromal stage were included among participants with low serum ergothioneine levels at baseline.
We are unable to specify which mushroom varieties were predominantly consumed by participants in the town of Hisayama.
Given the limited discriminative ability of serum ergothioneine and potential degradation due to long-term sample storage, we were unable to explore a clinically meaningful threshold value of serum ergothioneine.
Generalizability of findings was limited because participants of this study were recruited from one town in Japan.
These findings suggest that the potential benefit of ergothioneine may be attenuated in individuals with pre-existing, multifactorial risk profiles for dementia.
Our findings showed that higher serum ergothioneine levels were associated with a lower risk of developing all-cause dementia, AD, and non-AD dementia in an older Japanese population. Since ergothioneine cannot be synthesized in the human body, a diet rich in ergothioneine may be beneficial in reducing the risk of dementia.”
For five years I got most of my estimated 7 mg daily ergothioneine intake from mushrooms in AGE-less chicken vegetable soup per Ergothioneine dosing. The soup was always boring, but I got too bored this year and stopped making it. I haven’t replaced mushroom intake with supplements.
I still don’t eat fried or baked foods, preferring sous vide and braising cooking methods to avoid exogenous advanced glycation end products. I avoid buying foods that evoke a hyperglycemic response or otherwise form excessive endogenous AGEs per All about AGEs.
Continuing Part 1 with three 2025 papers, starting with a rodent study of dietary mussel plasmalogens’ effects on atherosclerosis:
“The purpose of this study was to clarify the underlying mechanisms of Mytilus edulis-derived plasmalogens (Pls) against atherosclerosis (AS) in ApoE−/− mice induced by a high-fat diet (HFD), through a comprehensive analysis of hepatic metabolomics and aortic transcriptomics data. Besides Pls role as the storage pool of n-3 PUFAs, the structural feature of vinyl ether bond at sn-1 position confers multiple advantages upon Pls compared to their diacyl counterparts, including enhanced antioxidant capacity, increased membrane fluidity, as well as improved stability and stability of biomembranes.
The C57BL/6 mouse strain is susceptible to high-fat diet (HFD)-induced AS lesions, and ApoE knockout accelerates AS development. Molecular mechanisms by which Pls ameliorate AS were investigated through a comprehensive analysis of hepatic metabolomics and aortic transcriptome profiles, focusing on changes in gene related to the p38 mitogen-activated protein kinase (MAPK) signaling pathway and the downstream inflammatory response.
The concentration of Pls in mussel tissues is 32 μgmg−1 (dry weight), and the obtained Pls contains 49.53% of phosphatidylethanolamine-Pls, 35.87% of phosphatidylcholine-Pls, and 14.60% of phosphatidylserine-Pls. The main fatty acid compositions of Pls are presented in Supplementary Table 1, which indicates that EPA accounts for 45.82% and the n-3/n-6 ratio is 3.84.
Pls inhibited aortic lipid accumulation, prevented thickening of the aortic wall, and suppressed collagen accumulation at the aortic-heart junction. Pls inhibited HFD-induced loosening of hepatocyte arrangement, vacuolization, and accumulation of lipid droplets.
Although several key components of MAPK signaling pathway were suppressed at both the transcriptional and protein levels in Pls-treated mice, no significant changes in phosphorylated p38 protein were observed among the experimental groups in our study. Further research is needed to elucidate the overall inhibitory mechanism of Pls on p38 protein and the MAPK signaling pathway.”
A rodent / human cell study investigated effects of plasmalogens in innate immune system macrophages on atherosclerosis:
“We demonstrate that simultaneous inactivation of two key enzymes involved in macrophage polyunsaturated fatty acid (PUFA) metabolism—ELOVL5, which elongates long-chain PUFAs, and LPCAT3, which incorporates them into phospholipids—disrupts membrane organization by promoting the formation of cholesterol-enriched domains. This increases macrophage sensitivity to cytotoxic oxysterols and leads to more vulnerable atherosclerotic plaques with enlarged necrotic cores in a mouse model of atherosclerosis.
We identified ELOVL5 as one elongase facilitating the conversion of C20 to C22 PUFA. In humans, analysis of 187 carotid plaques reveals a positive correlation between LPCAT3/ELOVL5-generated phospholipids—including arachidonate (C20:4 n-6)-containing ether lipids—and more stable plaque profiles. Additionally, Mendelian randomization analysis supports a causal relationship between LPCAT3 expression and reduced risk of ischemic stroke.
Potentially beneficial effects we observed in mice and in human atheroma plaques were mainly associated with PLs enriched in omega-6, particularly in AA. Although omega-6 FAs are often considered as pro-inflammatory, their role is undergoing reconsideration, with markers linked to the intake of omega-6 appearing beneficial in the context of cardiovascular diseases. In this context, it is worth to note that AA-containing plasmalogens have been previously identified as markers of healthy obesity.
Our findings uncover a regulatory circuit essential for PUFA-containing phospholipid generation in macrophages, positioning PUFA-containing ether lipids as promising biomarkers and therapeutic targets.”
A human study included plasmalogens in investigating associations among people with mental illness and their lipid profiles:
“Plasma lipidomic profiles of 623 individuals (188 schizophrenia (SCZ), 243 bipolar disorder (BD), 192 healthy controls) belonging to the PsyCourse Study were assessed using liquid chromatography and untargeted mass spectrometry. Exact etiology of these major mental health disorders is yet unknown and while their symptoms overlap, their diagnostic criteria are based on clinical evaluations of symptoms without objective markers.
Cognitive dysfunction is among the most disabling symptoms of SCZ and BD, and is difficult to treat with the commonly used pharmacologic regimes. Consequently, it has important impacts on long-term functional outcomes.
We aimed to answer the question, whether specific lipid species or classes were associated with differential performance across various cognitive domains, including psychomotor and processing speed, executive function, short-term and working memory and crystalized intelligence and whether these associations were affected by diagnoses.
Lipids belonging to the phosphatidylethanolamine plasmalogen (PE-P) class emerged as the main lipid class associated negatively with DG-SYM test performance, representative of processing and psychomotor speed. Our findings showed that higher levels of PE-P 42:5, PE-P 40:4, PE-P 40:5, and ceramide 38:1 in plasma samples of our study are significantly associated with poorer DG-SYM test performance. The DG-SYM test mainly measures processing speed, the amount of time required to complete a series of cognitive tasks. Enrichment analysis also showed significant associations between other lipid classes and various cognitive tests.
Our findings suggest a link between lipids and cognitive performance independent of mental health disorders. Independent replication is warranted to better understand if phosphatidylethanolamines could represent an actionable pharmacologic target to tackle cognitive dysfunction, an important unmet clinical need that affects long-term functional outcomes in individuals with severe mental health disorders.”
It was apparently beyond these researchers’ expertise to offer informed discussion on this study’s associative link between enrichment of these three phosphatidyl ethanolamine plasmalogens and cognitive dysfunction. Grok countered that their depletion was associated with neurodegenerative diseases (Alzheimer’s, Parkinson’s, multiple sclerosis), cardiovascular risk / oxidized-LDL burden, and chronic fatigue / post-viral syndromes.
Continuing Plasmalogens Week with two 2025 papers, starting with a rodent study of plasmalogens’ effects on mitigating cognitive decline:
“We evaluated beneficial effects of plasmalogens (PLS), phosphatidylcholine (PC), and phosphatidylserine (PS) on age-associated cognitive decline. We established a mouse model of aging-associated cognitive impairment using the subcutaneous injection of d-galactose (D-gal) at a dosage of 400 mg/kg/day.
We randomly divided six-week-old female mice into nine groups: control, model, high-dose PLS (0.3 mg/kg/day), low-dose PLS (0.09 mg/kg/day), high-dose PC (200 mg/kg/day), low-dose PC (50 mg/kg/day), high-dose PS (200 mg/kg/day), low-dose PS (50 mg/kg/day), AMC-Plas (120 mg/kg/day; and functional component PLS (0.252 mg/kg/day).
We administered PLS, PC, and PS separately by oral gavage once daily. We extracted PLS from scallops according to the literature. AMC-Plas is a commercially available health supplement known for its neuroprotective properties and memory-enhancing effects. In this study, we included AMC-Plas as a positive control group to evaluate the effects of different phospholipids.
Synaptophysin (SYP), synapsin-1 (SYN-1), postsynaptic density protein 95 (PSD-95), and brain-derived neurotrophic factor (BDNF) play important roles in synapse formation and synaptic plasticity. Synaptic function alterations or losses are key pathological mechanisms that underlie development of cognitive impairment. Therapeutic strategies that attempt to restore synaptic function or promote synaptic remodeling are considered to be increasingly promising strategies to mitigate cognitive decline.
Results showed that:
PLS improved spatial memory performance by 44% and object recognition by 80% in D-galactose-induced cognitively impaired mice.
PLS significantly decreased glial fibrillary acidic protein (GFAP)-positive cells (an indicator of astrocyte activation) in the dentate gyrus (DG) of the hippocampus, an important result because the DG is a crucial neurogenesis region.
PLS alleviated neuronal damage and protected against synaptic injury, verified by a 228% increase in PSD-95 expression in the hippocampus.
PLS showed a more prominent role for the mitigation of age-related cognitive impairment compared with PC and PS.
In conclusion, the evaluation of PLS using both behavioral and neuropathological assessments in cognitively impaired mice highlighted its exceptional efficacy compared with other phospholipids. PLS at a remarkably low effective dose significantly ameliorated cognitive deficits in cognitively impaired mice. This result further emphasized its potential relevance in neurodegenerative disease research.
We found that PLS alleviated cognitive impairment potentially by improving synaptic function; however, the molecular mechanisms that underlie its effects on synaptic function warrant further investigation.”
There was no disclosed chemical analysis of the PLS scallop extract’s plasmalogen types or other contents. Despite its name, I didn’t see that the AMC-Plas product contained plasmalogens or plasmalogen precursors.
A fruit fly study investigated plasmalogen effects on mitochondria during aging:
“We identify plasmalogens—endogenous ether-linked phospholipids—as key regulators of age-associated mitochondrial fission in Drosophila melanogaster. Loss of Kua (also known as plasmanylethanolamine desaturase (PEDS) / TMEM189 in mammals), the enzyme essential for plasmalogen biosynthesis, leads to inhibition of mitochondrial fission and impaired recruitment of the fission protein Drp1, similar to what is observed during aging.
Mitochondrial dynamics, comprising balanced cycles of fission and fusion, are essential for preserving organelle quality, metabolic flexibility, and cellular homeostasis throughout life. Aging disrupts this balance, with multiple studies reporting a decline in mitochondrial fission that contributes to the accumulation of enlarged and dysfunctional mitochondria.
These morphological changes are linked to impaired mitophagy, altered energy production, and tissue dysfunction. Midlife induction of Drp1—the dynamin-related GTPase that drives mitochondrial division—has been shown to reverse age-related mitochondrial defects and prolong lifespan in Drosophila.
To determine whether plasmalogen biosynthesis is essential for mitochondrial fission, we used KuaMI04999, a hypomorphic allele. Western blot analysis revealed significantly reduced Kua protein levels in KuaMI04999/+ heterozygotes compared to wild-type controls.
Our findings reveal a previously unrecognized lipid-based mechanism that controls mitochondrial fission during aging and position plasmalogens as key effectors linking membrane composition to mitochondrial homeostasis. It is not merely expression or stability of Drp1 that is affected, but rather its recruitment to the mitochondrial surface, which is a critical activation step for fission.
While our study highlights the requirement of plasmalogen biosynthesis for Drp1 recruitment, further work is needed to understand how plasmalogens mechanistically facilitate this interaction.”
A 2025 rodent study investigated mechanisms by which erythropoietin (EPO) enables adult neurogenesis and cognitive function:
“We mapped epigenomic and transcriptional landscapes of adult mouse hippocampus under recombinant human EPO (rhEPO) treatment. We discovered significant lineage-specific remodelling of chromatin accessibility predominantly in newly formed pyramidal neurons, highlighting a robust EPO-driven neurogenic response. Notably, transposable elements (TEs), particularly ancient LINEs and SINEs, emerged as critical cis-regulatory elements (cCREs).
EPO is known to be upregulated in the brain under hypoxic or injury conditions, and it has been considered a natural neuroprotective agent. We demonstrated that EPO, a traditionally hematopoietic hormone, can profoundly reprogram the adult neural epigenome to drive neurogenesis.
EPO may activate a specific subclass of dormant regulatory elements to drive nearby genes. Such a mechanism would represent a previously unappreciated mode of gene regulation: the de novo recruitment of ancient genomic elements to drive a contemporary cellular response.
Our data support the model that EPO drives differentiation of progenitors rather than inducing widespread cell division. The net effect is an enrichment of pyramidal neurons at the cost of interneurons. Pyramidal neurons integrate in the hippocampal circuitry, leading to potential implications for mood, memory, cognitive enhancement, and recovery from brain injury.
We propose a conserved evolutionary mechanism at play: ancient TEs embedded in the genome have been repurposed as cCREs in neural cells, and during an EPO-induced neurogenic stimulus, the brain taps into this reservoir of regulatory elements to rapidly reshape gene expression. In evolutionary terms, this represents an efficient strategy.”
A 2025 reply to a letter to the editor cited 56 references to elaborate on Part 1 and related topics:
“A positive effect does not necessarily mean benefit, and positive effects on individual organisms may mean adverse effects on other coexisting organisms. However, a vast literature shows that hormetic stimulation can result in benefits depending on the context, for instance, clear growth, yield, and survival improvement.
There is some energetic cost to support hormetic stimulation, with a likely positive energy budget, which might also have negative consequences if there is insufficient energy substrate, especially under concurrent severe environmental challenges. Moreover, hormetic preconditioning could be particularly costly when there is a mismatch between the predicted environment and the actual environment the same individuals or their offspring might face in the future.
Hormesis should not be unilaterally linked to positive and beneficial effects without considering dose levels. For any research to answer the question of whether a stimulation represents hormesis and whether it is beneficial, robust dose–response evaluations are needed, which should be designed a priori for this purpose, meeting the requirements of the proper number, increment, and range of doses.
Both additivity and synergism are possible in the hormetic stimulatory zone, depending also on the duration of exposure and the relative ratio of different components. This might happen, for example, when a chemical primes stress pathways (e.g., heat shock proteins and antioxidants), thus enabling another chemical to trigger hormesis (defense cross-activation) and/or because combined low subtoxicity may modulate receptors (e.g., aryl hydrocarbon receptor and nuclear factor erythroid 2-related factor 2) differently than individual exposures (receptor binding synergy).
Moreover, even when stimulation occurs in the presence of individual components, stimulation may no longer be present when combined, and therefore, effects of mixtures cannot be accurately predicted based on the effects of individual components. There may be hormesis trade-offs; hormesis should be judged based on fitness-critical end points.
While often modeled mathematically, hormesis is fundamentally a dynamic biological process and should not be seen as a purely mathematical function, certainly not a linear one. Much remains to be learned about the role of hormesis in global environmental change, and an open mind is needed to not miss the forest for the trees.”
“Hormetic-based interventions, particularly priming (or preconditioning), do not weaken organisms but strengthen them, enhancing their performance and health under different environmental challenges, which are often more massive than the priming exposure.
The catabolic aspect of hormesis is primarily protective whereas the anabolic aspect promotes growth, and their integration could optimize performance and health. The concept of preconditioning has also gained widespread attention in biomedical sciences.”
Reference 40 was a 2021 review that characterized hormesis as a hallmark of health:
“Health is usually defined as the absence of pathology. Here, we endeavor to define health as a compendium of organizational and dynamic features that maintain physiology.
Biological causes or hallmarks of health include features of:
Spatial compartmentalization (integrity of barriers and containment of local perturbations),
Maintenance of homeostasis over time (recycling and turnover, integration of circuitries, and rhythmic oscillations), and
An array of adequate responses to stress (homeostatic resilience, hormetic regulation, and repair and regeneration).
Disruption of any of these interlocked features is broadly pathogenic, causing an acute or progressive derailment of the system.
A future ‘medicine of health’ might detect perilous trajectories to intercept them by targeted interventions well before the traditional ‘medicine of disease’ comes into action.”
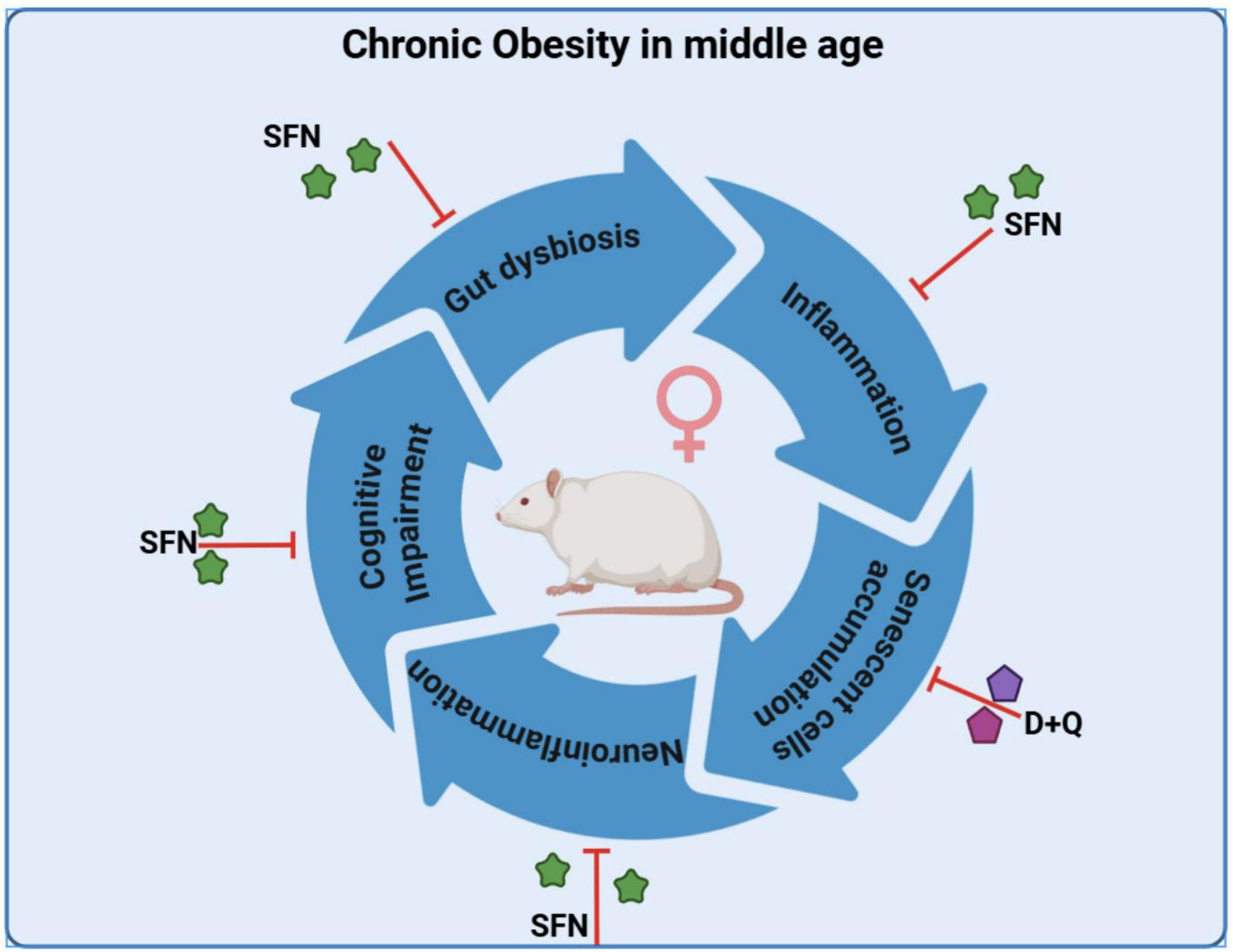
A 2025 rodent study by the same group as Part 1 investigated similar subjects from a different experimental angle of senotherapy effects on brain and behavior rather than cardioprotective effects of dasatinib / quercetin (a senolytic combination) and sulforaphane (senomorphic):
“This is the first study to analyze the effect of senotherapy in the brain of a model of chronic obesity in middle-aged female rats. D + Q reduced the pro-inflammatory cytokines evaluated in the obesity model. It did not improve memory and learning nor the expression of molecules associated with the maintenance of synapses.
In contrast, sulforaphane (SFN), which without eliminating senescent cells, decreased pro-inflammatory factors, increased IL-10, as well as brain-derived neurotrophic factor BDNF, synaptophysin (SYP), and postsynaptic density protein 95 (PSD-95), which, in turn, were associated with an improvement in behavioral tests in obese rats. This suggests that modulating the senescence-associated secretory phenotype (SASP), rather than eliminating senescent cells, might have better effects.”
A 2025 rodent study investigated effects of far-infrared light on Alzheimer’s disease models. I’ll focus on its Nrf2 findings:
“Far-infrared radiation (FIR) is commonly utilized as a complementary treatment of a range of disease, for example, insomnia and rheumatoid arthritis. In this research, we explored how FIR light impacts cognitive functions of TgCRND8 AD mice and elucidated its underlying molecular mechanism.
Infrared radiation is a form of electromagnetic energy that has wavelengths between 750 nm and 1000 μm, which are longer than visible light. International Commission on Illumination categorizes infrared light as three sub-divisions according to the wavelength: (1) near-infrared radiation (0.7–1.4 μm), (2) middle infrared radiation (1.4–3.0 μm), and (3) far-infrared radiation (3.0–1000 μm).
Nrf-2/ HO-1 signaling, a key endogenous antioxidant system, helps mitigate oxidative stress and enhances expression of various endogenous genes. Activation of HO-1 during inflammatory conditions may serve as an adaptiveresponse to reduce cytotoxicity through various mechanisms.
In this study, we applied EFFIT LITE® as the FIR spectrum transmitter which stably radiates an FIR spectrum with a wavelength of 4–20 μm, and the device was put within 1 cm directly above the head of the 3-month-old TgCRND8 mice for 30 min exposure once every day. FIR light notably enhanced cognitive function and spatial memory of TgCRND8 mice after 28-days consecutive treatment.
Underlying molecular mechanisms involve suppression of Aβ deposition, hyperphosphorylation of tau, and neuroinflammation through modulating Jak-2/Stat3 and Nrf-2/HO-1 pathways. Our current experimental findings amply indicate that FIR light is a potential non-pharmacological therapy for AD.”
This study measured Nrf2 and its quickly-induced downstream enzyme HO-1 effects of daily far-infrared light exposure for 30 minutes. We’d have to see measurements of Nrf2’s more-slowly induced and longer-lasting downstream xenobiotic detoxifying enzyme NQO1 to compare far-infrared light Nrf2 activation effects with those of natural plant compounds.
A 2025 human study investigated effects of long-term exercise:
“Exercise has well-established health benefits, yet its molecular underpinnings remain incompletely understood. We conducted an integrated multi-omics analysis to compare effects of acute vs. long-term exercise in healthy males.
Acute exercise induced transient responses, whereas repeated exercise triggered adaptive changes, notably reducing cellular senescence and inflammation and enhancing betaine metabolism. Exercise-driven betaine enrichment, partly mediated by renal biosynthesis, exerts geroprotective effects and rescues age-related health decline in mice.
Betaine binds to and inhibits TANK-binding kinase 1 (TBK1), retarding the kinetics of aging.
Betaine effectively alleviated senescence phenotypes by reduced senescence-associated β-galactosidase (SA-β-Gal)-positive cells, decreased p21 expression, lowered DNA damage indicator γ-H2A.X, and elevated heterochromatin mark H3K9me3. Betaine treatment also enhanced cellular antioxidant capacity, as evidenced by increased NRF2 phosphorylation and reduced ROS accumulation.
These findings systematically elucidate the molecular benefits of exercise, and position betaine as an exercise mimetic for healthy aging.”
A 2025 review subject was sulforaphane and brain health. This paper was the latest in a sequence where the retired lead author self-aggrandized his career by citing previous research.
He apparently doesn’t personally do what these research findings suggest people do. The lead author is a few weeks older than I am, and has completely white hair per an interview (Week 34 comments). I’ve had dark hair growing in (last week a barber said my dark hair was 90%) since Week 8 of eating broccoli sprouts every day, which is a side effect of ameliorating system-wide inflammation and oxidative stress.
If the lead author followed up with what his research investigated, he’d have dark hair, too. Unpigmented white hair and colored hair are both results of epigenetics.
Contrast this lack of personal follow-through of research findings with Dr. Goodenowe’s protocol where he compared extremely detailed personal brain measurements at 17 months and again at 31 months. He believes enough in his research findings to personally act on them, and demonstrate to others how personal agency can enhance a person’s life.
It’s every human’s choice whether or not we take responsibility for our own one precious life. I’ve read and curated on this blog many of this paper’s references. Five years ago for example: