People will forgive you for being wrong, but they will never forgive you for being right – especially if events prove you right while proving them wrong. Thomas Sowell
A 2026 study investigated aging through the use of transcriptomics:
“By constructing transcriptomic clocks of expected mortality across >11,000 samples from mouse, rat, macaque and human, we provide a unified framework that integrates age-associated transcriptional change with the direction and magnitude of lifespan modulation by interventions, enabling a quantitative and biologically interpretable readout of health status.
While chronological clocks captured many detrimental conditions (e.g., chronic diseases or certain short-lived genetic models), they were comparatively less responsive to lifespan-extending interventions, consistent with reports that longevity treatments often only modestly reverse age-associated transcriptomic signatures. In contrast, mortality clocks robustly distinguished both short- and long-lived models and showed strong associations with human time-to-death, approaching the predictive performance of second-generation DNAm mortality clocks while remaining biologically interpretable.
Our results support and extend DNAm-based observations that heterochronic parabiosis in old animals and early embryogenesis can induce molecular age deceleration. In old heterochronic parabionts, mortality clocks detected a sustained tAge reduction that persisted for 2 months after detachment from young partners. Together, these findings support the view that early development contains a conserved rejuvenation-like phase, and that systemic environment can partially reset molecular age later in life.
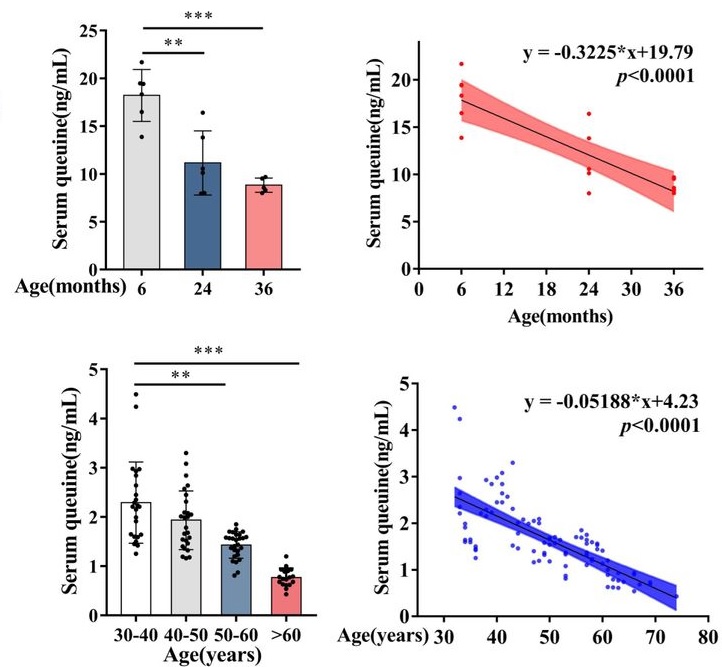
“The contribution of transfer RNA (tRNA)-specific modifications to aging remains largely unexplored. We systematically profile tRNA modifications across multiple organs, species, and senescence models, and identify mannosyl-queuosine (manQ) as the first tRNA-specific modification that consistently declines with age.
Across species, queuine supplementation extends lifespan and enhances healthspan. In naturally aging mice, long-term oral administration beginning at 16-months-old (human equivalent 50 years) extends mean lifespan by 15.3%, reduces DNA methylation age, improves cognitive and motor performance, strengthens antioxidant defenses, remodels the gut microbiota, and alleviates inflammation and metabolic dysfunction without detectable toxicity.
These findings establish tRNA epitranscriptomic remodeling as a previously unrecognized layer of aging regulation, and identify restoration of manQ through queuine supplementation as a multi-system strategy to delay aging.
manQ hypomodification is selective rather than reflecting global tRNA depletion. Aging preferentially reduces the manQ-containing tRNAAsp fragment while leaving the corresponding unmodified tRNAAsp fragment, and other queuosine-modified tRNAs, relatively unchanged.
This pattern supports a regulated defect in modification homeostasis rather than a generalized change in transcript abundance. Such specificity argues that manQ loss is not merely a passive consequence of tissue degeneration, but instead represents a conserved, biologically meaningful aging-associated event with mechanistic impact.
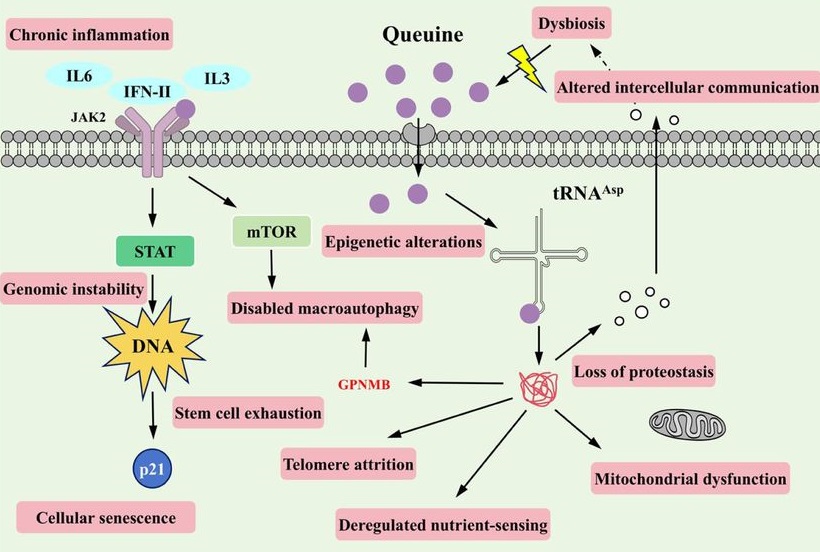
Because proteostasis intersects with multiple canonical hallmarks (e.g. mitochondrial dysfunction, impaired stress resilience, and altered intercellular communication), translation-coupled proteome destabilization offers a unifying explanation for how a single tRNA modification defect can elicit multi-system consequences. In this view, manQ decline is not merely one of many molecular changes observed in aging, but rather a proximate determinant capable of amplifying downstream hallmarks through a common axis of proteome quality control.
Our findings further suggest that manQ depletion may engage self-reinforcing feedback loops that accelerate aging trajectories. This architecture offers a conceptual framework in which aging progressively erodes ‘epitranscriptomic integrity’ at the tRNA level, pushing translation toward an error-prone regime that accelerates proteostatic collapse and functional decline.
A distinctive implication of this work is that queuine introduces a microbiota-host epitranscriptomic axis into aging biology. Queuine is produced by gut microbiota and cannot be synthesized de novo by mammals. These findings expand the conceptual scope of geroscience by placing a microbiota-derived nutrient upstream of translational quality control.
Queuine supplementation offers a distinct therapeutic logic: rather than modulating a single signaling cascade, it restores a tRNA modification state that governs translational fidelity – an upstream determinant of proteome quality that can, in principle, influence multiple downstream hallmarks concurrently. These findings highlight an intervention paradigm centered on restoring molecular fidelity, rather than suppressing a single downstream phenotype, as a strategy to delay systemic aging.”
A 2026 primate study investigated effects of vitamin C:
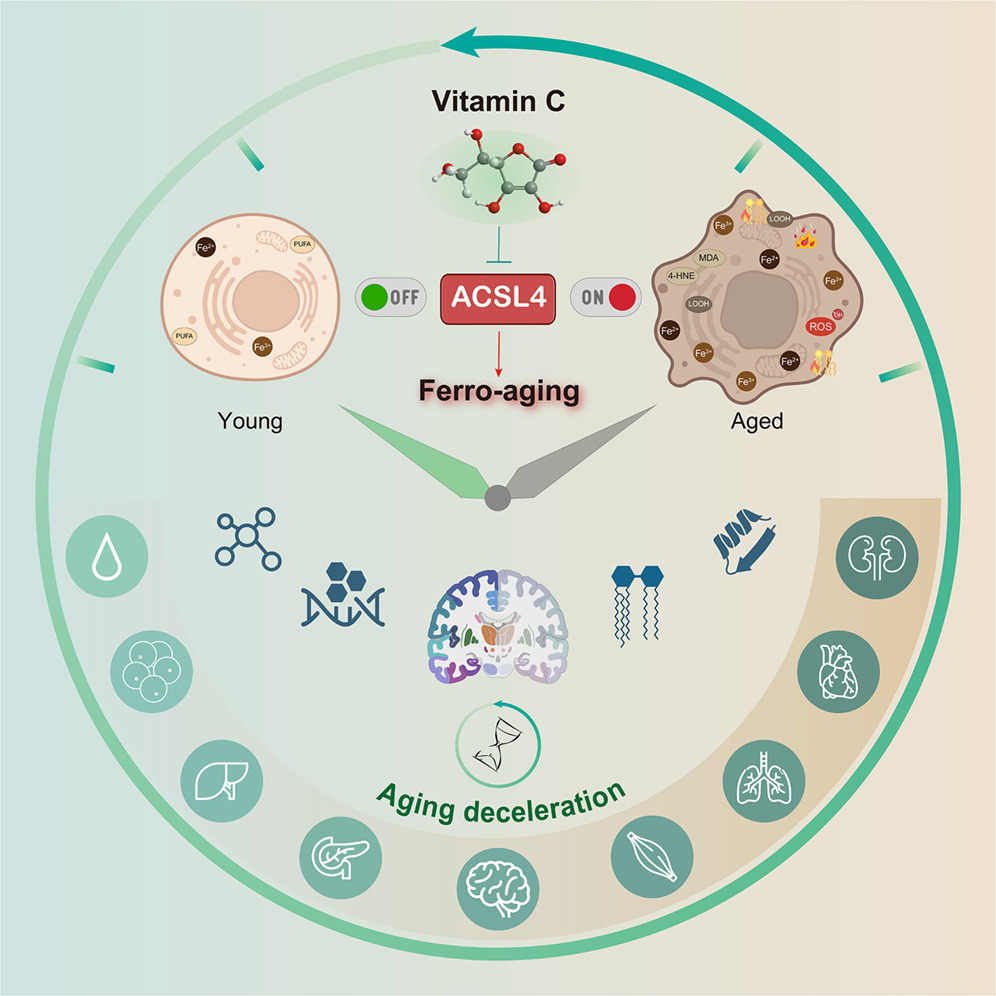
“Here, we define a conserved iron-lipid axis driving primate aging, termed ‘ferro-aging.’ Multi-tissue profiling in humans and non-human primates reveals age-progressive iron accumulation, fueling chronic lipid peroxidation orchestrated by acyl-coenzyme A (CoA) synthetase long-chain family member 4 (ACSL 4). Distinct from acute ferroptosis, this ACSL4-mediated process promotes cellular senescence and systemic functional decline.
We identify vitamin C (VC) as a direct inhibitor of ACSL4. Long-term VC administration in aged monkeys for over 40 months potently reduces ferro-aging signatures across tissues, attenuates multi-organ pathology, and improves neurological and metabolic functions. Multi-omic aging clocks indicate the VC-mediated reversal of biological age.
Despite decades of interest in oxidative stress, largely sparked by the free radical theory of aging, efforts to modulate it broadly with antioxidants have yielded inconsistent or neutral outcomes, highlighting the theory’s limitations and underscoring the need to identify more specific, upstream drivers. A critical challenge remains: determining whether the iron-lipid axis constitutes a core upstream driver of aging in primates and, if so, whether it is therapeutically targetable.
In this study, we bridge these gaps. We define an iron-triggered, ACSL4-governed, lipid peroxidation-driven program that escalates with age across diverse cell types and multiple organs in non-human primates.
VC treatment dose-dependently increased Nrf2 phosphorylation and activation. VC orchestrates a dual-defense strategy against ferro-aging: it directly suppresses the pro-aging lipid peroxidation driver ACSL4, while in parallel, it bolsters the cell’s intrinsic antioxidant capacity via Nrf2 pathway activation.
Middle-aged cynomolgus monkeys (12–16 years old, approximating human 40–50 years) received daily oral VC (30 mg/kg group) or a control treatment for 40 months under standardized conditions.
Structural MRI analysis demonstrated that VC intervention counteracted age-related brain atrophy. Using general linear mixed models, we found that VC restored cortical surface area in the frontal lobes of aged monkeys. Regional analysis identified enlargement in four regions of the orbital frontal cortex, an area critical for adaptivebehavior.
Diffusion MRI-based connectomics revealed that, compared with young animals, aged monkeys exhibited reduced structural connectivity in 18 brain regions. VC treatment restored connectivity in 9 of these regions, which were predominantly located in the posterior parietal cortex, a hub for spatial awareness and decision-making.
VC exerted robust neuroprotective effects. It attenuated heterochromatin loss (increased H3K9me3) in the prefrontal cortex and hippocampus and reduced abnormal protein aggregates, including cytosolic aggresomes and Aβ. Additionally, VC lowered the abundance of activated microglia and astrocytes and suppressed expression of the innate immune sensor cGAS in the hippocampus.
VC supplementation reduced the estimated biological age across multiple organs. At the epigenetic level, VC lowered DNA methylation age in several tissues, including brain, brown adipose tissue, muscle, skin, aorta, and kidney. In the hippocampus, the most substantial reductions in biological age occurred in microglia, oligodendroglia, and oligodendrocyte precursor cells. In the pancreas, alpha cells, beta cells, and ductal cells showed the greatest rejuvenation.
In summary, chronic VC supplementation inhibits the ferro-aging pathway, reduces multidimensional biological age across primate organs, and ameliorates a spectrum of functional declines in nervous and metabolic systems. Our work establishes ACSL4 inhibition as a promising and translationally relevant therapeutic strategy for mitigating aging-related decline.
A long-term, 40-month intervention study in aged non-human primates is a highly translational model given their shared inability with humans to synthesize VC endogenously. The finding that a single, safe nutrient can reverse multidimensional aging clocks in a primate has profound implications for translational longevity medicine.”
“For humans (who, like macaques, cannot synthesize vitamin C), the Recommended Dietary Allowance (RDA) is 75–90 mg/day for adults (~1–1.5 mg/kg for a 60–70 kg person) to prevent deficiency. Upper safe intake levels are much higher: up to 2,000 mg/day (Tolerable Upper Intake Level) is considered safe for most adults, with no established adverse effects at that level from food/supplements.
Treated monkeys represent advanced aging stages (likely equivalent to human 50s–70s+ based on ‘aged’ designation and long-term intervention effects), extending the prior 12–16-year monkey range (human ~35–55) to broader anti-aging applications. While human trials are needed, the primate evidence (long-duration, systemic benefits) strengthens the case for high-dose, sustained vitamin C as a strategy against ferro-aging in humans. It elevates vitamin C from a nutrient to a targeted anti-aging compound in primates.”
Coincidentally, I started taking extra vitamin C separately from other supplements in the form of liposomal 1 gram twice daily this past winter. Can’t say that it had any effects on my intended target, avoiding sniffles and sneezing, as allergy season kicked off in early February. With this study’s findings, I’ll continue.
A 2026 review subject was mechanisms and therapeutic potential for Nrf2 activators in combination with mesenchymal stem cells:
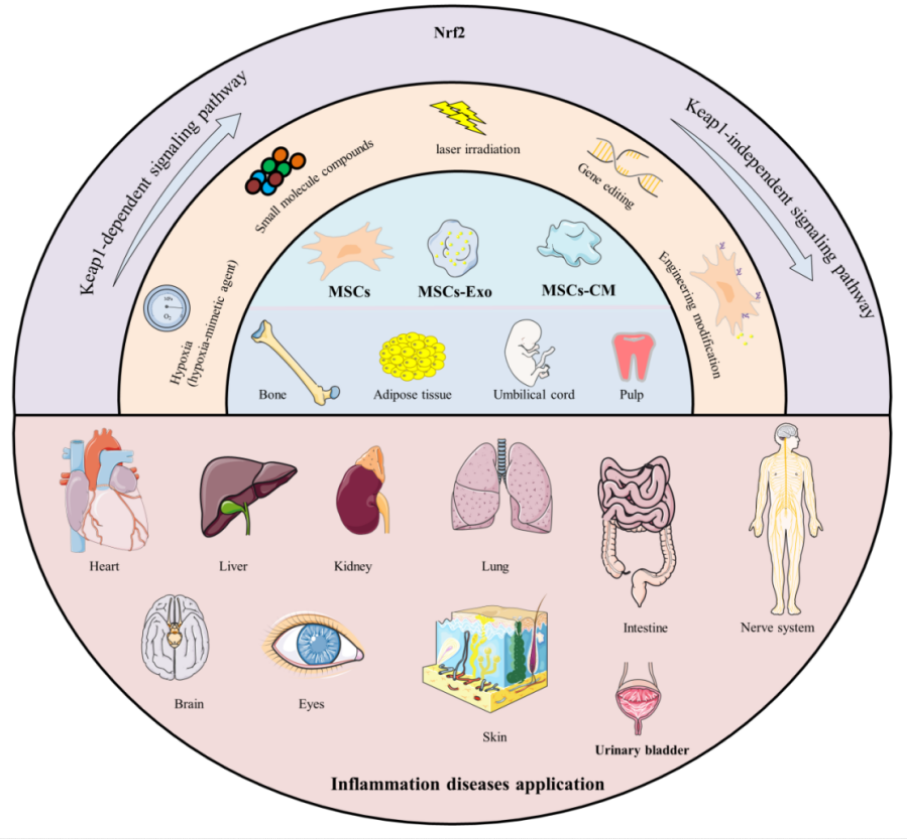
“Mesenchymal stromal/stem cells (MSCs) are multipotent stem cells that can be isolated from various tissues – such as bone marrow (BM), umbilical cord (UC), adipose tissue (AD), dental pulp (DP), hair follicle (HF), and placenta – and differentiated into multiple lineages under appropriate conditions. Their functional repertoire includes immunomodulation, homing, and differentiation, which collectively help establish a balanced inflammatory and regenerative niche within damaged tissues during severe inflammation. MSCs-derived extracellular vesicles (MSCs-EVs) and conditioned medium (MSCs-CM) play remarkable roles, exhibiting potent anti-inflammatory and antioxidant properties that offer novel therapeutic alternatives for inflammatory diseases.
Therapeutic capacity of MSCs in inflammatory conditions is increasingly attributed to their potent paracrine activity rather than solely to their differentiation potential. A key mechanism underlying this paracrine effect is activation of the Nrf2 antioxidant pathway.
MSCs and their secreted products including exosomes (Exos), EVs, and CM, activate Nrf2 through multi-dimensional/target mechanisms, thereby enhancing cellular antioxidant defenses, modulating immune responses, and promoting tissue repair. It is noteworthy that therapeutic efficacy of MSCs and their derivatives can be enhanced through external modulation, including pretreatment with natural compounds.
Preconditioning refers to brief treatment of MSCs or their derivatives with physical, chemical, or biological factors prior to application, aiming to enhance their ability to counteract oxidative stress and improve their therapeutic efficacy. Flavonoids precondition and prime MSCs via the direct Keap1-Nrf2 pathway or indirect PI3K-Akt pathway, which enhances cellular resilience to adverse conditions by reducing apoptosis and promoting survival. Primed MSCs, in turn, remodel the microenvironment through an altered secretory profile, releasing bioactive factors that create more favorable conditions for their own persistence.
The core logic of these strategies lies in simulating or inducing adaptive stress, such as employing specific chemical molecules or drug stimuli, or utilizing physical / microenvironmental preconditioning to mimic specific physical conditions of the in vivo injury environment. The most straightforward strategy is overexpression of Nrf2 or its key downstream effector molecules.
The majority of existing studies remain at the level of observing correlations with Nrf2 upregulation, and there is still a lack of precise causal validation regarding key upstream signals – such as specific cytokines, miRNAs, or proteins – through which MSCs or derivatives initiate Nrf2 activation. Mechanistic insights are predominantly derived from in vivo or rodent (mouse/rat) model experiments, with a notable absence of clinical validation, insufficient long-term safety and pharmacokinetic data, and a lack of standardization in administration routes and dosages, all of which hinder clinical translation.
The essential role of the Nrf2 pathway has not been rigorously confirmed, as most studies have not employed reverse genetic validation using Nrf2-knockout animals or specific inhibitors. Consequently, it remains unclear whether therapeutic effects are necessarily and exclusively dependent on Nrf2, and potential synergistic contributions from other pathways may have been overlooked.
Most natural flavonoids face challenges such as low oral bioavailability, rapid metabolism, and poor targeting. Numerous challenges remain to be addressed in order to translate these promising preclinical findings into clinical practice. Future research should focus on the following aspects:
Elucidating precise upstream molecular mechanisms by which MSCs activate Nrf2;
Employing more clinically relevant chronic disorder models;
Systematically evaluating long-term safety, optimal delivery strategies (including dosage and route of administration), and immunogenicity of MSCs-based therapies;
Validating selection criteria (optimal source), quality control, batch-to-batch consistency of MSCs, and addressing regulatory and ethical barriers to clinical translation; and
Integrating molecular docking, ADMET (Absorption, Distribution, Metabolism, Excretion, Toxicity) prediction, and in vitro and in vivo validation to further elucidate regulatory effects of flavonoids and enhance understanding of their mechanisms of action.”
This paper was overly long at 127 pages, so I focused on the later sections. None of these treatments are currently ready for clinical trials.
I also didn’t mention specific flavonoids as Nrf2 activators. It’s beyond a reviewer’s task to rank Nrf2 activators, and a study’s researchers seldom address why they used a poorly-activating flavonoid instead of a higher-ranked natural plant compound such as sulforaphane.
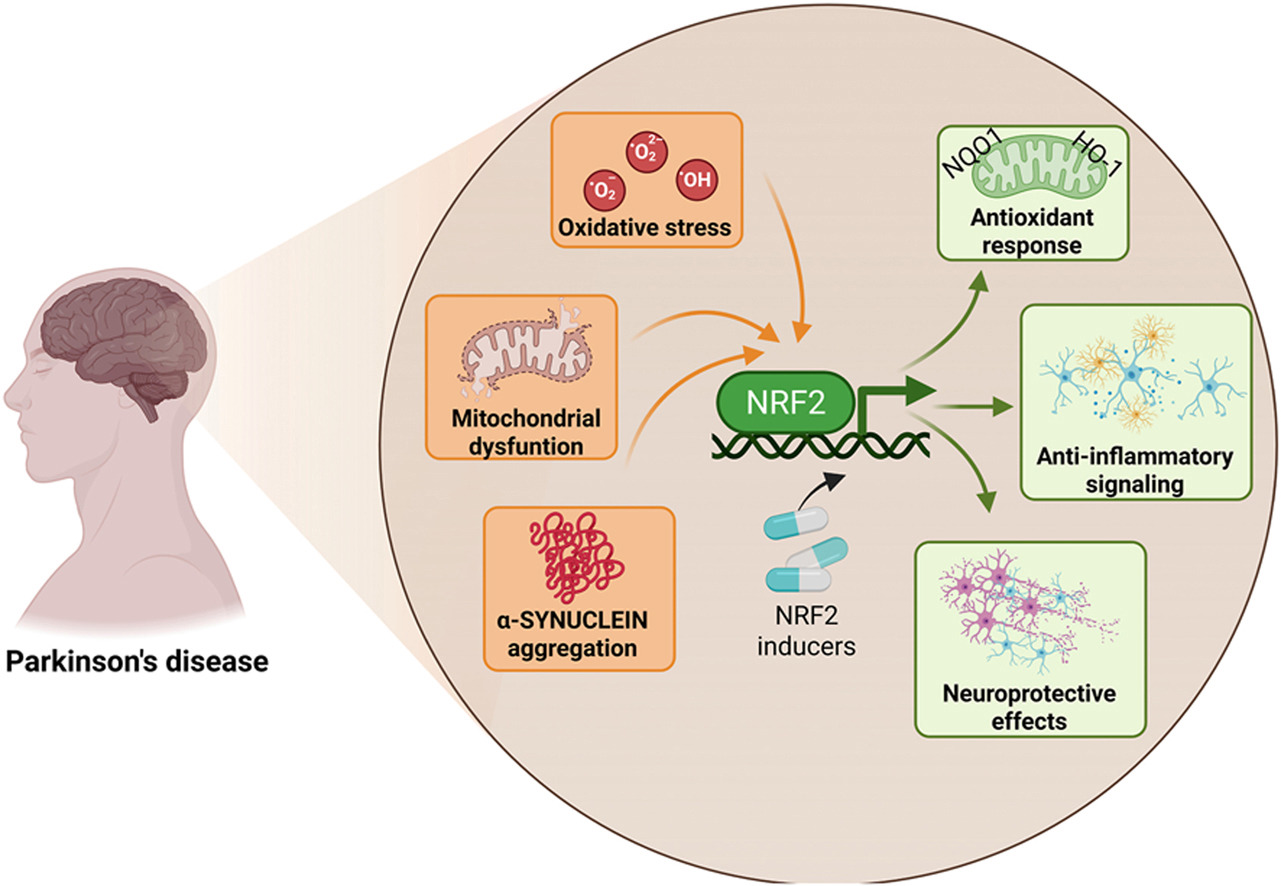
Starting this blog’s twelfth year by curating a poorly-done 2026 review of Nrf2 and its capability to change a person’s development of Parkinson’s disease. I’ll emphasize precedent conditions that if not effectively dealt with in youth, can’t prevent PD from occurring at some later life stage.
“This review explicitly examines how age-associated decline in NRF2 responsiveness intersects with redox imbalance, mitochondrial dysfunction, proteostatic failure, and neuroinflammation, core mechanisms shared between aging and PD. PD unfolds through a complex interplay of cellular stress and immune responses. Oxidative stress, mitochondrial dysfunction, and chronic neuroinflammation converge to damage dopaminergic neurons, with microglia playing a central role in amplifying this injury.
NRF2 emerges as a key regulator of antioxidant defenses, inflammatory balance, and mitochondrial protection, offering a promising target for clinical intervention. NRF2 activity favors the anti-inflammatory microglial over the pro-inflammatory phenotype. Decline in NRF2 inducibility with age impairs microglial clearance, promotes neuroinflammation, and reduces antioxidant defenses, while NRF2 activation restores protective functions and offers a promising therapeutic target.
Strategies aimed at restoring or enhancing NRF2 activity hold significant promise as disease-modifying interventions, not only to slow PD progression but also to promote resilience against the broader spectrum of age-associated neurodegenerative and inflammatory conditions.”
This review only gave lip service to PD progression outside of the brain, as if the importance of prodromal factors to a person’s neurodegeneration such as dysfunction in gut, eyes, skin, and olfactory systems can be minimized. But failure to recognize early what will doom a person to be unable to recover health in later decades is disingenuous. Since these reviewers omitted early interventions into PD prodromal factors, the best they came up with was interventions to “slow PD progression.”
Maybe these reviewers felt it would be outside the scope of this review to discuss early non-brain PD factors for more than one sentence? However, while PD is defined by striatal brain neurons, Nrf2 activity is much less in brain and central nervous system neurons than elsewhere in the body per Nrf2 Week #2: Neurons.
I disagree with these reviewers’ self-imposed emphasis on aging. Repeating ‘age-associated’ numerous times seemed as if they wanted to influence the reader into thinking age in and of itself was a cause for PD, rather than an imputed mathematical correlation. Their emphasis led to dumb mentions such as senolytics for no apparent reason than senescence is a ‘hallmark of aging’, and to meaningless ‘diseasome of aging’ characterizations, and to ignoring the existence of early non-age-associated PD diagnoses in 20- and 30-year-olds.
Whatever it takes to get published, I’d guess. Or maybe it’s that the number of omissions and useless points a review paper makes increases with the number of reviewers and their sponsors’ agendas.
For example, why was it permissible to dedicate lip service to ‘exposome’ factors like microplastics, environmental pollution, and viruses, but it’s still not permitted in 2026 to discuss research into the impacts on vascular disease and neurodegeneration of lipid nanoparticles and DNA contamination in what a large number of humans were exposed to by injected pharmaceuticals starting in late 2020? Not to mention two studies published in 2024 of over 2.5 million people whose incidences of neurologic issues, mild cognitive impairment, and Alzheimer’s disease rapidly increased after ‘vaccination’?
I’ve mentioned in this blog many times how it’s every human’s choice whether or not we take responsibility for our own one precious life. I suggest, if it’s not too late, do that for your children’s lives, too.
Here are five 2025 human ergothioneine studies, starting with a clinical trial of healthy older adults:
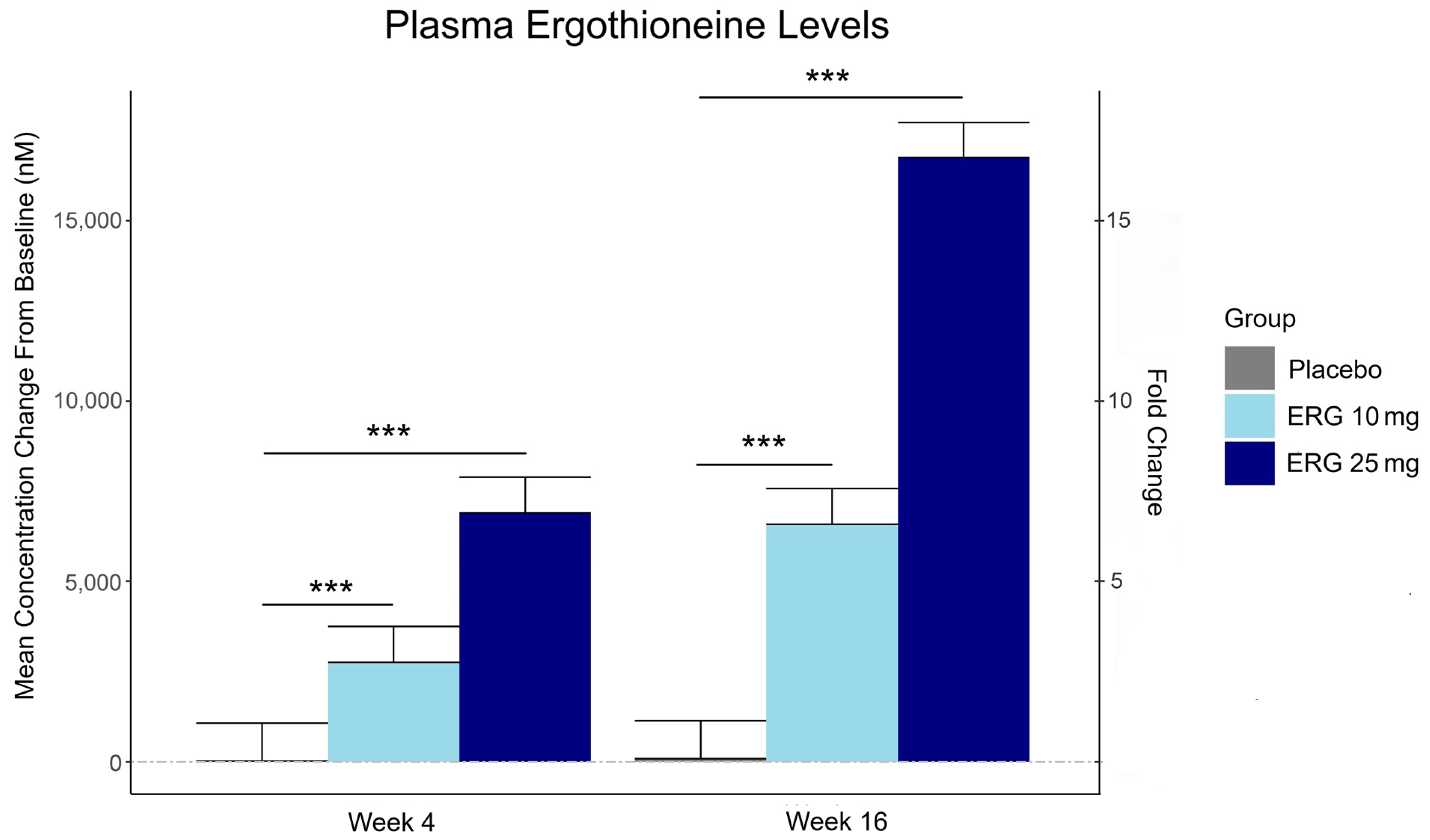
“In this 16-week randomized, double-blind, placebo-controlled trial, 147 adults aged 55–79 with subjective memory complaints received ergothioneine (10 mg or 25 mg/day ErgoActive®) or placebo. Across all the groups, approximately 73% of participants in each group were female, with a median age of 69 years.
The primary outcome was the change in composite memory. Secondary outcomes included other cognitive domains, subjective memory and sleep quality, and blood biomarkers. At baseline, participants showed slightly above-average cognitive function (neurocognitive index median = 105), with plasma ergothioneine levels of median = 1154 nM.
Although not synthesized in the human body, ergothioneine is efficiently absorbed via the OCTN1 transporter (also known as the ergothioneine transporter, or ETT), which is expressed in many tissues, including the intestine, red blood cells, kidneys, bone marrow, immune cells, skin, and brain. This transporter enables ergothioneine to accumulate in high concentrations in organs vulnerable to oxidative stress and inflammation. Ergothioneine has multiple cellular protective functions, including scavenging reactive oxygen species, chelating redox-active metals, suppressing pro-inflammatory signaling, and protecting mitochondrial function.
Plasma ergothioneine increased by ~3- and ~6-fold for 10 mg, and ~6- and ~16-fold for 25 mg, at weeks 4 and 16, respectively.
While the primary outcome, composite memory, showed early improvement in the 25 mg group compared to baseline, this effect was not sustained and did not differ from placebo. Reaction time showed a significant treatment-by-time interaction favoring ergothioneine, yet the between-group differences were not significant, suggesting that any potential benefits were modest and require validation in larger or longer studies.
Other cognitive effects observed were primarily within-group and not consistently dose-responsive, highlighting the challenge of detecting objective cognitive changes over a relatively short study duration in high-functioning healthy populations. However, positive effects of ergothioneine supplementation were observed on subjective measures of prospective memory and sleep initiation that were not seen in the placebo group.
This trial adds to the growing body of evidence supporting the favorable safety profile of ergothioneine. No adverse events attributable to ergothioneine were reported. Additionally, we observed potential hepatoprotective effects, with significant reductions in the plasma AST and ALT levels, particularly among males in the ERG 25 mg group.”
https://www.mdpi.com/1661-3821/5/3/15 “The Effect of Ergothioneine Supplementation on Cognitive Function, Memory, and Sleep in Older Adults with Subjective Memory Complaints: A Randomized Placebo-Controlled Trial”
The third graphic for Ergothioneine dosing, Part 2 showed a human study where a 25 mg dosing stopped after Day 7, but the plasma ergothioneine level stayed significantly higher than baseline through Day 35.
The second graphic for Ergothioneine dosing, Part 2 was a male mouse experiment where plasma ergothioneine levels of a human equivalent 22 mg to 28 mg daily dose kept rising through 92 weeks.
This trial couldn’t explain the desirability of a 25 mg daily dose that was likely (per the second and third graphics for Ergothioneine dosing, Part 2) to sustain the subjects’ increased plasma ergothioneine levels well after the trial ended at Week 16. What effects can be expected from a sustained plasma ergothioneine level that’s 16 times higher than the subjects’ initial levels? Were these 16-fold sustained plasma ergothioneine levels better or worse than the 6-fold increases in the 10 mg group, both of which were likely to continue past the trial’s end?
A representative of the trial’s sponsoring company talked a little more about the trial in this interview:
Another clinical trial investigated ergothioneine’s effects on skin:
“We conducted an 8-week, randomized, double-blind, placebo-controlled clinical trial to evaluate effects of daily oral supplementation with 30 mg of ergothioneine (DR.ERGO®) on skin parameters in healthy adult women aged 35–59 years who reported subjective signs of skin aging. Objective measurements including melanin and erythema indices, skin glossiness, elasticity, and wrinkle and pigmentation counts were used to comprehensively evaluate changes in skin condition.
The OCTN1 transporter is preferentially expressed in basal and granular epidermal layers, where cellular renewal and barrier maintenance are most active. Once internalized, ergothioneine localizes to mitochondria, where it directly scavenges reactive oxygen species (ROS) and protects mitochondrial DNA from UV- and inflammation-induced damage.
At the signaling level, ergothioneine activates key protective pathways such as the Nrf2/ARE axis, enhancing expression of antioxidant enzymes including HO-1, NQO1, and γ-GCLC. These enzymes contribute to redox homeostasis and glutathione regeneration, reinforcing cellular defense systems against photoaging and environmental insult.
In parallel, ergothioneine modulates the PI3K/Akt/Nrf2 and SIRT1/Nrf2 pathways, which are implicated in collagen preservation, inflammation resolution, and mitochondrial maintenance. These pathways converge to reduce matrix metalloproteinase (MMP) activity, enhance collagen synthesis, and suppress pro-inflammatory cytokines (TNF-α, IL-6, IL-1β), all of which are central to maintaining skin structure and function.
Compared to placebo, the DR.ERGO® ergothioneine group showed significantly greater improvements in melanin and erythema reduction, skin glossiness, elasticity, and wrinkle and spot reduction. No adverse events were reported.
These findings corroborate and extend previous clinical evidence from (Hanayama et al., 2024), who investigated an ergothioneine-rich mushroom extract (Pleurotus sp., 25 mg ergothioneine/day) in a 12-week randomized double-blind trial, and (Chunyue Zhang, 2023), who examined pure ergothioneine supplementation (25 mg/day) in a 4-week open-label study. We contextualized our results within this existing literature by comparing key outcomes.
Several limitations should be acknowledged:
The study cohort consisted solely of Japanese women aged 35–59 years, which may limit generalizability across sexes, ethnicities, and age groups.
The 8-week intervention period, while sufficient to detect short-term effects, does not allow conclusions about the sustainability of benefits or the risk of relapse upon discontinuation.
The placebo group also showed modest improvements in self-perception, highlighting the well-documented placebo response in beauty and wellness studies.
This study focused on a single daily dosage (30 mg/day) without evaluating dose–response relationships, and hydration-specific endpoints such as corneometry or transepidermal water loss (TEWL) were not included.”
Two clinical trials investigated ergothioneine’s effects on sleep quality:
“A four-week administration of 20 mg/day ergothioneine (EGT), a strong antioxidant, improves sleep quality; however, its effect at lower doses remains unclear. This study estimated the lower effective doses of EGT using a physiologically based pharmacokinetic (PBPK) model in two clinical trials.
In Study 1, participants received 5 or 10 mg/day of EGT for 8 weeks, and their plasma and blood EGT concentrations were measured. An optimized PBPK model incorporating absorption, distribution, and excretion was assembled. Our results showed that 8 mg/day of EGT for 16 weeks was optimal for attaining an effective plasma EGT concentration.
In Study 2, a randomized, double-blind, placebo-controlled study, participants received 8 mg/day EGT or a placebo for 16 weeks. The subjective sleep quality was significantly improved in the EGT group than in the placebo group.
In mammals, EGT is not generated in the body but is acquired from the diet via the carnitine/organic cation transporter OCTN1/SLC22A4. Its plasma concentration after oral administration is quite stable and gradually increases after repeated dosing on a multi-day basis.
Blood concentrations of EGT increase after Day 8 when EGT intake is interrupted, and they continue to increase until Day 35. The delayed increase in EGT concentration in the blood, compared with that in the plasma, can be interpreted as its efficient uptake by undifferentiated blood cells, which express high levels of OCTN1/SLC22A4 in the bone marrow, and subsequent differentiation to mature blood cells that enter the circulation. This may imply the nonlinear absorption, distribution, and excretion of EGT owing to saturation of the transporter at higher concentrations, potentially leading to difficulty in model construction.
This is the first study to propose a strategy to estimate lower effective doses based on the PBPK model.”
The bolded section above referenced a 2016 study / third graphic for Ergothioneine dosing, Part 2, where a 25 mg dosing stopped after Day 7, but the plasma ergothioneine level stayed high through Day 35. I didn’t see that the referenced 2004 and 2010 studies addressed this 2016 finding.
I also didn’t see that this study’s mathematical model accounted for saturation of the OCTN1 transporter or other effects, such as a very small ergothioneine clearance rate. Okay, lower the ergothioneine dose, and achieve a lower persistent plasma ergothioneine level, to what benefit?
“The present study demonstrated that OCTN1 is associated with myeloid cells rather than lymphoid cells, and especially with erythroid-lineage cells at the transition stage from immature erythroid cells to peripheral mature erythrocytes.”
Persistent high ergothioneine levels aren’t costless. Skewing bone marrow stem cells and progenitor cells toward a myeloid lineage is done at the expense of lymphocytes, T cells, B cells, and other lymphoid lineages.
Where are the studies that examine these tradeoffs? Subjective sleep quality in this study and sleep initiation in the first study above aren’t sufficiently explanatory.
A study investigated associations of plasma ergothioneine levels and cognitive changes in older adults over a two-year period:
“Observational studies have found that lower plasma levels of ergothioneine (ET) are significantly associated with higher risks of neurodegenerative diseases. However, several knowledge gaps remain:
Most of the above studies were based on cross-sectional study design, and potential reverse causation cannot be excluded. It has been suggested that plasma ET declines concomitantly with the deterioration of cognitive function.
Since the impact of a single dietary factor on health is mild, it is prone to be affected by the baseline characteristics of subjects (such as sex, educational level, disease status and gene polymorphism). However, no study has systematically evaluated potential effect modifiers on the association between ET levels and cognitive function.
The dose-response distribution between ET and cognitive function remains undetermined.
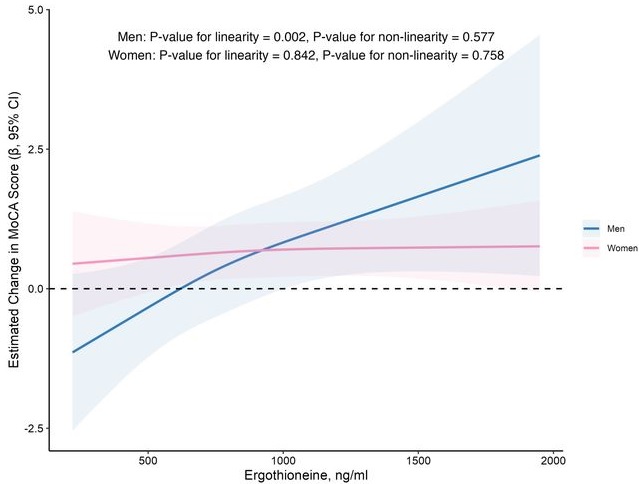
In this prospective cohort study of 1,131 community-dwelling older adults (mean age 69 years), higher baseline plasma ET levels were significantly associated with slower cognitive decline, as assessed by Montreal Cognitive Assessment (MoCA) scores, during a 2-year follow-up period.
When the plasma concentration of ET exceeds 1,000 ng/mL, the decline in cognitive function significantly slows down. However, this association has only been observed in men.
Domain-specific analysis found that the observed ET-MoCA association was mainly driven by the temporary slowdown in the decline of visuospatial/executive and delayed recall. Impaired delayed recall represents one of the earliest and most sensitive cognitive markers of dementia progression, predictive of conversion from MCI to dementia. The preferential preservation of this function by ET suggests targeted neuroprotective effects within the hippocampus.
Visual inspection of the spline curves revealed a potential plateauing effect at ET concentrations ≥1,000 ng/mL in the total population.
Baseline ET concentrations differed between men and women. Most men (81.5%) had concentrations below 1,000 ng/mL (median 754.2, IQR 592.0–937.9 ng/mL). Women exhibited substantially higher median plasma ET concentrations than men, with 35.7% of women exceeded 1,000 ng/mL (median 890.1, IQR 709.7–1,095.6 ng/mL).
Our study included only participants with normal cognitive function, and the results remained robust even after excluding those with baseline cognitive function at the lower end of the normal range. Collectively, our findings support that low ET intake occurs prior to cognitive decline.
Our findings indicate that higher plasma ET levels are significantly associated with slower cognitive decline independent of confounders in non-demented community-dwelling elderly participants, with such association observed in men but not women. Dose-response curves indicated plateauing effects above 1000 ng/mL.”
The average age of this study and the first trial above were both 69 years. Since the first trial’s participants showed slightly above-average cognitive function (neurocognitive index median = 105), with plasma ergothioneine levels of median = 1154 nM at baseline, and this study showed plateauing effects above 1000 ng/mL, I wonder how raising plasma ergothioneine levels above 1000 ng/mL could possibly show a net benefit for older people? What are the trade-offs for older people between potentially increasing slightly above-average cognitive function with ergothioneine and its other effects from saturating their OCTN1 transporter?
This study is at its preprint stage. I’m interested to see if its peer review prompts these researchers to also investigate the common finding that people who are most deficient at baseline have the greatest improvements. If so, would these sex-specific associations still hold?
Wrapping up with a study that investigated associations of serum ergothioneine levels with the risk of developing dementia:
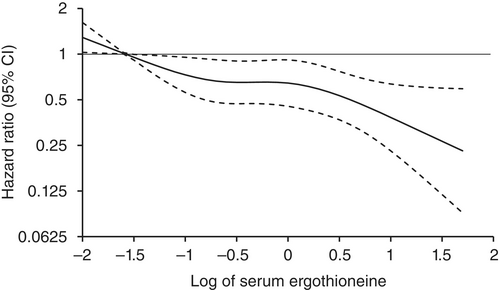
“1344 Japanese community-residents aged 65 years and over, comprising 765 women and 579 men, without dementia at baseline were followed prospectively for a median of 11.2 years.
During follow-up, 273 participants developed all-cause dementia. Among them, 201 had Alzheimer’s disease (AD) and 72 had non-Alzheimer’s disease (non-AD) dementia.
Age- and sex-adjusted hazard ratios (HRs) for all-cause dementia, AD, and non-AD dementia decreased progressively across increasing quartiles of serum ergothioneine. These associations remained significant after adjustment for a wide range of cardiovascular, lifestyle, and dietary factors, including daily vegetable intake.
In subgroup analysis, association between serum ergothioneine levels and the risk of dementia tended to be weaker in older participants and in women:
In older individuals, cumulative burden of multiple risk factors such as hypertension, diabetes mellitus, and smoking may contribute to both neurodegenerative and vascular pathology, potentially diminishing the relative influence of ergothioneine.
In women, postmenopausal hormonal changes, particularly the decline in estrogen, have been associated with increased oxidative stress and a higher vulnerability to neurodegenerative changes.
Several limitations should be noted:
Since serum ergothioneine levels and other risk factors were measured only at baseline, we could not evaluate the changes of serum ergothioneine levels during the follow-up period. Lifestyle modifications during follow-up could have influenced serum ergothioneine levels and other risk factors. In addition, serum ergothioneine level was measured only once, and from a sample.
We cannot rule out residual confounding factors, such as other nutrients in mushrooms and socioeconomic status.
There is a possibility that dementia cases at the prodromal stage were included among participants with low serum ergothioneine levels at baseline.
We are unable to specify which mushroom varieties were predominantly consumed by participants in the town of Hisayama.
Given the limited discriminative ability of serum ergothioneine and potential degradation due to long-term sample storage, we were unable to explore a clinically meaningful threshold value of serum ergothioneine.
Generalizability of findings was limited because participants of this study were recruited from one town in Japan.
These findings suggest that the potential benefit of ergothioneine may be attenuated in individuals with pre-existing, multifactorial risk profiles for dementia.
Our findings showed that higher serum ergothioneine levels were associated with a lower risk of developing all-cause dementia, AD, and non-AD dementia in an older Japanese population. Since ergothioneine cannot be synthesized in the human body, a diet rich in ergothioneine may be beneficial in reducing the risk of dementia.”
For five years I got most of my estimated 7 mg daily ergothioneine intake from mushrooms in AGE-less chicken vegetable soup per Ergothioneine dosing. The soup was always boring, but I got too bored this year and stopped making it. I haven’t replaced mushroom intake with supplements.
I still don’t eat fried or baked foods, preferring sous vide and braising cooking methods to avoid exogenous advanced glycation end products. I avoid buying foods that evoke a hyperglycemic response or otherwise form excessive endogenous AGEs per All about AGEs.
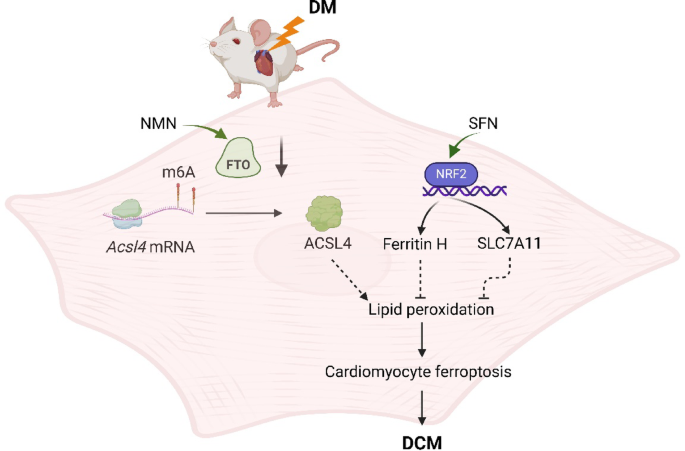
A 2025 rodent study investigated synergistic effects of sulforaphane (SFN) and nicotinamide mononucleotide (NMN) on diabetic cardiomyopathy:
“Diabetic cardiomyopathy (DCM) as a significant diabetes complication remains a major human challenge. In this study, we provide evidence that the fat mass and obesity-associated protein (FTO) plays a pivotal role in DCM pathogenesis.
Downregulation of FTO in DCM acts as a critical inducer of ferroptosis by increasing expression of acyl-CoA synthetase long-chain family 4 (ACSL4), a key positive mediator of ferroptosis. FTO-mediated mitigation of ferroptosis occurs in an ACSL4-dependent manner which leads to increased methylation of Acsl4 transcripts.
Ferroptosis plays an essential role in the pathogenesis of DCM.
As the most widespread mRNA modification, N6-methyladenosine (m6A) is globally downregulated and implicated in diabetes and its complications.
FTO, which is an m6A demethylase, was found to be downregulated in diabetes and its cardiovascular complications.
NAD+ enhances the demethylase activity of FTO. Dietary supplementation with NMN, a critical intermediate in the NAD+biosynthetic pathway, has been shown to efficiently elevate endogenous NAD+ levels.
Enhancing the demethylase activity of FTO with NMN combined with SFN targeting NRF2 could synergistically reduce the level of lipid peroxides to inhibit ferroptosis, providing an effective avenue for alleviating DCM.
We found that NMN could alleviate ferroptosis and improve heart function through enhancing FTO. SFN could prevent ferroptosis and partly rescue heart function via AMPK-mediated NRF2 activation.
We demonstrated that SFN combined with NMN treatment could significantly inhibit lipid peroxidation and rescue cardiac function in DCM compared to SFN or NMN treatment alone.
Although the combined regimen further suppressed ferroptosis and improved cardiac performance, it fell short of complete remission, underscoring that additional pathways also contribute substantially to the pathogenesis of DCM.”
The epigenetic mechanism involved with this study’s dietary dissolved-in-water 100mM NMN dose was Non-CpG methylation. This study used the same very low sulforaphane dose intraperitoneally injected as Eat broccoli sprouts for your heart. Discussion of that study provided an example that if a person waited until a diabetes-related disease condition became a problem, capabilities to adequately address causes and prevent the problem may be lost.
Notice in the last bar of the second graphic above taken from Figure 7 that the combined treatment was also provided to non-diabetic mice. These researchers provided over a dozen other measurements in Figure 7 to show similar short-term non-effects of the combined treatment, i.e. that it neither benefited nor harmed non-diabetic subjects. Grok interpreted this study’s 3-month-long intervention to be a 1-to-5 year human equivalent, depending on the measured effect (shorter for metabolic effects like MDA, longer for structural cardiac changes like reduced ferroptosis.)
The male subjects began at 2-months old, a human-equivalent 15-20 years old. These researchers gave them diabetes by feeding them a “high-fat diet for 3 months to induce insulin resistance, followed by a single intraperitoneal injection of streptozotocin (STZ) (in 0.1 mol/L of citrate acid buffer, 60 mg/kg) to induce partial insulin deficiency.” A 5-months old mouse is a 25-30 years old human equivalent.
Grok considered this study’s NMN human equivalent dose to be extremely high if provided in drinking water, but not if injected, depending on volume. However, the study didn’t state that its NMN dose was injected, and there was no dose volume indicated.
Here are two 2025 papers, starting with a rodent study that investigated interactions between the Nrf2 and kynurenine pathways:
“Exposure to the tryptophan metabolite kynurenine and its electrophilic derivative kynurenine-carboxyketoalkene (Kyn-CKA) leads to an increase in the abundance of transcription factor Nrf2 and induction of Nrf2-target genes. The Keap1/Nrf2 system is the main orchestrator of cellular defence against environmental stress, most notably oxidative and inflammatory stress.
Nrf2 can be activated pharmacologically by small molecules, the majority of which are electrophiles and oxidants that modify specific cysteine-based sensors in Keap1. C151 in Keap1 is the target of the isothiocyanate sulforaphane, a classical Nrf2 activator that has been employed in ∼90 clinical trials, as well as for the two Nrf2 activators that are clinically in use: dimethyl fumarate, for relapsing remitting multiple sclerosis, and omaveloxolone, for Friedreich’s ataxia.
Kynurenine is an endogenous metabolite derived from the essential amino acid tryptophan. Kynurenine and its metabolites, such as the electrophilic kynurenine-carboxyketoalkene (Kyn-CKA), have been demonstrated to activate Nrf2 in other pathologies, including sickle cell disease, attenuating inflammation. Moreover, identification of the gene encoding the kynurenine-metabolising enzyme kynureninase as a gene transcriptionally upregulated by Nrf2, provides a plausible negative feedback regulatory mechanism.
Because kynurenine is not electrophilic, whereas its metabolite Kyn-CKA is, we considered the possibility that Kyn-CKA is the actual Nrf2 activator. Using biochemical and cell-based assays, we found that Kyn-CKA reacts with C151 in the BTB domain of Keap1 and increases the thermostability of Keap1, indicating target engagement. Consequently, Nrf2 accumulates and induces transcription of antioxidant/electrophile-responsive element (ARE/EpRE)-driven genes.
These findings demonstrate that Kyn-CKA targets C151 in Keap1 to derepress Nrf2, and reveal that Nrf2 is a main contributor to the anti-inflammatory activity of Kyn-CKA in macrophages.”
A review subject was targeting nicotinamide adenine dinucleotide, oxidized form (NAD+) for clinical use:
“Mammalian NAD+ biosynthesis includes four known pathways, primarily occurring in cytoplasm:
(a) the NRH pathway;
(b) the salvage pathway;
(c) the Preiss–Handler pathway; and
(d) the kynurenine pathway.
The de novo kynurenine pathway metabolizes tryptophan (Trp) to NAD+, producing various intermediates that serve as biomarkers for different diseases. These intermediates show alterations in various pathological conditions.
While kynurenine and its metabolic derivatives are intermediates in the de novo NAD+ biosynthesis pathway, these are also produced independently in various physiological contexts, particularly in immune cells, where they act as immunomodulatory compounds.”
This second paper above showed a graphic of the Nrf2 and kynurenine pathways together in a diagram showing relationships between NAD+ augmentation and the hallmarks of aging, but didn’t elaborate other than labeling their box Dysbiosis. So how these two pathways interact is better outlined in the first paper above with explaining how a kynurenine-metabolizing enzyme is one of the hundreds of Nrf2 target genes, creating a natural feedback loop between Nrf2 activation and the kynurenine pathway.
These reviewers also lumped SIRT1 in their Dysbiosis box, and into several other boxes, probably due to the penultimate coauthor’s influence:
However, repeating something over and over doesn’t make it scientifically valid regardless of the number of citations. Or, as a 2022 review Sirtuins are not conserved longevity genes concluded:
“A global pursuit of longevity phenotypes was driven by a mixture of framing bias, confirmation bias, and hype. Review articles that propagate these biases are so rampant that few investigators have considered how weak the case ever was for sirtuins as longevity genes.
Acknowledging that a few positive associations between sirtuins and longevity have been identified after thousands of person-years and billions of dollars of effort, we review the data and suggest rejection of the notions that sirtuins (i) have any specific connection to lifespan in animals and (ii) are primary mediators of the beneficial effects of NAD repletion.”
A 2025 rodent study investigated dynamics of organ and circulating nicotinamide:
“Liver-derived circulating nicotinamide from nicotinamide adenine dinucleotide (NAD+) catabolism primarily feeds systemic organs for NAD+ synthesis. We surprisingly found that, despite blunted hepatic NAD+ and nicotinamide production in liver-specific nicotinamide nucleotide adenylyltransferase 1 (NMNAT1) deletion mice (liver-specific knockout [LKO]), circulating nicotinamide and extra-hepatic organs’ NAD+ are unaffected.
Metabolomics reveals a massive accumulation of a novel molecule in the LKO liver, which we identify as nicotinic acid riboside (NaR). The liver releases NaR to the bloodstream, and kidneys take up NaR to synthesize NAD+ through nicotinamide riboside kinase 1 (NRK1) and replenish circulating nicotinamide.
Serum NaR levels decline with aging, whereas oral NaR supplementation in aged mice boosts serum nicotinamide and multi-organ NAD+, including kidneys, and reduces kidney inflammation and albuminuria. The liver-kidney axis maintains systemic NAD+ homeostasis via circulating NaR, and NaR supplement ameliorates aging-associated NAD+ decline and kidney dysfunction.
While this study provides evidence of hepatic production and renal consumption of NaR for NAD+ homeostasis in mice, future human works are warranted to confirm these findings. In addition, genetic studies will be necessary to fully understand NaR metabolism at cellular and organismal levels.
While this study shows the oral availability of NaR and its effect on systemic NAD+ metabolism in mice, human studies testing NaR safety, oral availability, pharmacokinetics, and pharmacodynamics should be performed to test potential clinical usage of NaR supplements. Additionally, future studies are needed to investigate physiological significance of NT5C2-mediated hepatic production of NaR in healthy mice and identify NaR transporter(s).”
An elaborating commentary was published along with this study:
“Nicotinamide (NAM), nicotinamide riboside (NR), nicotinic acid (NA), and NAR are the salvageable precursors that feed into production of nicotinamide mononucleotide (NMN) and nicotinic acid mononucleotide (NAMN) to regenerate NAD coenzymes. NAMN is at an interesting juncture in NAD metabolism because it is formed in de novo synthesis and in salvage synthesis from both NA and NAR.
Song and coworkers did not specifically set out to determine endogenous sources of NR and/or NAR. Rather, they wanted to see what would happen when they deleted the major Nmnat isozyme, Nmnat1, in liver.
With depression of hepatic NAD+, they saw elevation of liver NMN and NAMN and discovered a huge increase in hepatic and circulating NAR. By viral knockdown, the step of conversion of accumulated NAMN to NAR was found to be catalyzed by a 5′ – nucleotidase encoded by the Nt5c2 gene, and the major tissue receiving the NAR was found to be the kidney.
Further, they showed that levels of NAR decline in aging while provision of supplementary NAR supports a newfound ability of the mouse kidney to circulate NAM. Of potential translational significance, supplementary NAR also supported mouse kidney function in aging.”
Continuing Part 1, here are four more 2025 human studies of the Nrf2 activator astaxanthin, starting with a randomized, double-blind, placebo-controlled trial of its effects on reducing oxidative stress and inflammatory responses following eccentric exercise:
“This study investigated effects of astaxanthin supplementation on plasma MDA and HMGB1 levels following acute eccentric exercise in recreationally active male students. Fifty-four students were assigned to receive either 12 mg/day of natural astaxanthin (AST, n = 27) or placebo (PLA, n = 27) for 14 days.
A key consequence of eccentric-induced muscle damage is overproduction of reactive oxygen species (ROS). When ROS production exceeds the capacity of endogenous antioxidant systems, lipid peroxidation can occur. Malondialdehyde (MDA) is a stable end-product of lipid peroxidation and serves as a widely recognized biomarker for oxidative stress and cell membrane damage.
In parallel, muscle cell damage results in release of damage-associated molecular patterns (DAMPs) into the extracellular space. Among these, High Mobility Group Box-1 (HMGB1) plays a central role in inflammation when passively released from the nucleus. HMGB1 acts as a potent pro-inflammatory signal by activating innate immune receptors, recruiting immune cells, and upregulating cytokines such as IL-6 and TNF-α.
This heightened immune activity contributes to delayed-onset muscle soreness, which typically peaks 24–72 hours post-exercise, and is associated with impaired recovery. Sustained elevations in oxidative and inflammatory biomarkers, including MDA and HMGB1, may further impair recovery and contribute to long-term muscle pathology.
Astaxanthin’s antioxidant effects are mediated through both direct and indirect mechanisms. Structurally, astaxanthin is a xanthophyll carotenoid with a unique polar–nonpolar–polar configuration that enables it to span the phospholipid bilayer of cell membranes. This positioning allows it to neutralize ROS both at the membrane surface and within the lipid bilayer.
In addition, astaxanthin enhances endogenous antioxidant defenses by upregulating enzymes such as superoxide dismutase (SOD), catalase, and glutathione peroxidase (GPx) through activation of the Nrf2–ARE signaling pathway. This dual mode of action provides both immediate and sustained protection against oxidative stress during and after exercise.
The placebo group showed substantial increases in MDA and HMGB1 after exercise, whereas the astaxanthin group experienced attenuated rises (~22% and ~27% smaller, respectively) and faster recovery toward baseline within 24 hours. These findings suggest that astaxanthin supplementation can be incorporated into recovery strategies for athletes and active individuals, especially during periods of heavy training or repeated bouts of intense eccentric exercise. By reducing oxidative damage and inflammation, astaxanthin may shorten recovery time, limit performance loss, and support overall training adaptations—benefits that are particularly valuable in sports requiring frequent high-intensity efforts.
Several limitations should be acknowledged in this study.
Sample size was relatively small and limited to recreationally active young males, which may restrict generalizability of findings to other populations such as females, older adults, or elite athletes.
Supplementation period was limited to 14 days; although this duration is sufficient to achieve plasma saturation of astaxanthin, longer interventions may produce different or more pronounced effects.
Only two biomarkers were assessed (MDA and HMGB1), which provide important but incomplete insights into broader oxidative stress and inflammatory response. Including additional markers such as enzymatic antioxidants, cytokine profiles, and muscle damage indicators (e.g., creatine kinase) could yield a more comprehensive understanding.
Dietary intake and physical activity outside the intervention were self-reported and not strictly controlled, which may have introduced variability in results.”
https://tmfv.com.ua/journal/article/view/3664/1922 “Taking Astaxanthin Supplementation Attenuates MDA and HMGB1 Following Eccentric Exercise: A Randomized Controlled Trial in Recreationally Active Students”
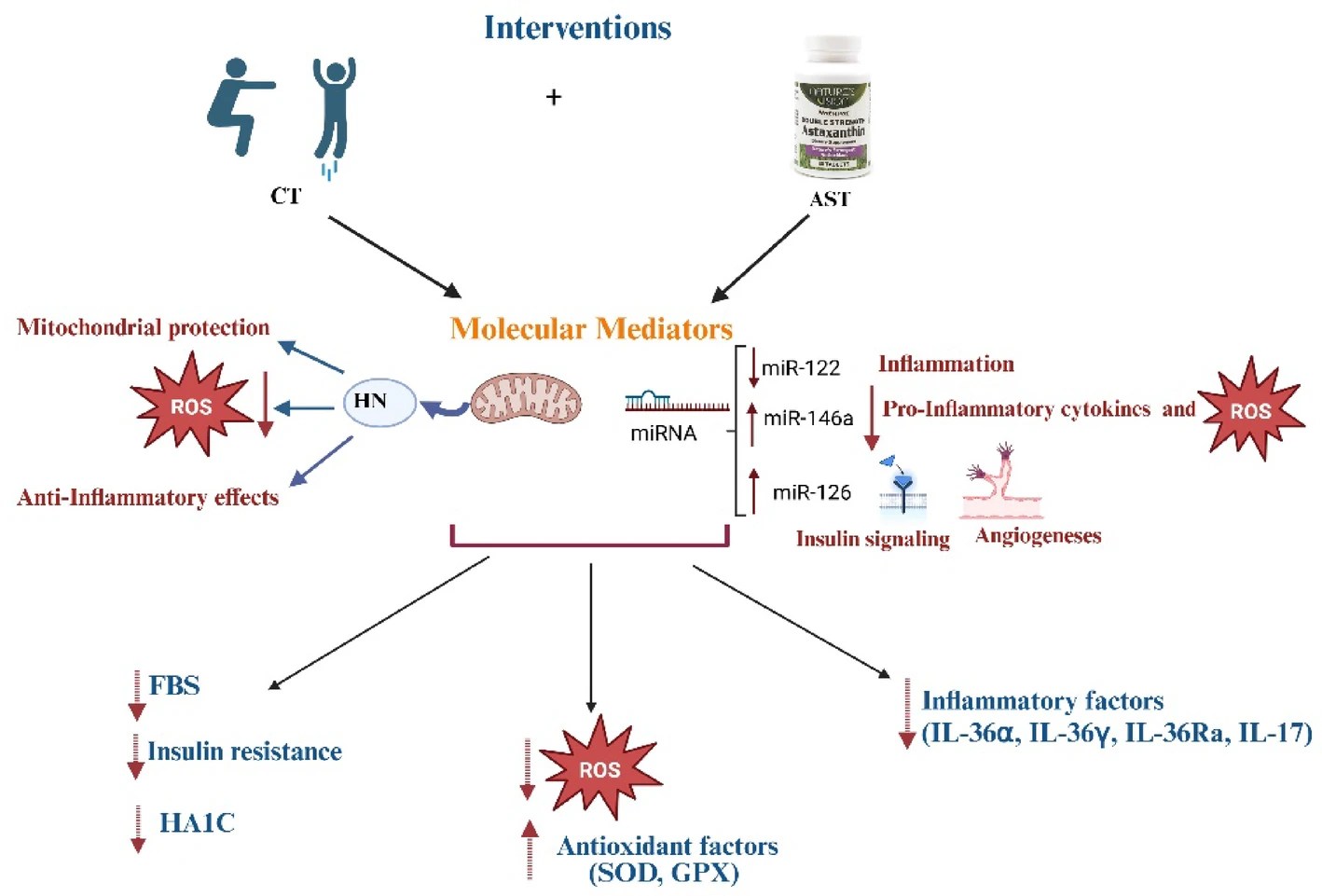
A clinical trial investigated astaxanthin’s effects with exercise in diabetic women:
“This study examined whether combined aerobic and resistance training (CT) and astaxanthin (AST) supplementation synergistically improve oxidant and inflammatory status as well as metabolic indices in T2DM, focusing on the mediatory role of Humanin (HN) and microRNAs (miRNA-122, miRNA-126-3p, and miRNA-146a).
Ninety women with T2DM were randomly assigned to six groups (n = 15 each):
Control (C), placebo (P), AST supplementation (S), combined training (CT), CT + placebo (CT + P), and CT + AST supplementation (CT + S).
CT, CT + P and CT + S groups underwent an 8-week training program (eight exercises, three sessions per week).
S and CT + S groups received 8 mg/day of AST.
This study only enrolled female participants age between 30 and 60 years old to minimize inter-individual biological variability arising from sex differences in hormone regulation, fat distribution, and gene expression related to inflammation and oxidative stress. Oxidative stress (OS) markers, inflammatory cytokines, HN levels, miRNAs expression, fasting blood glucose (FBG), insulin resistance (HOMA-IR), lipid profile, and hemoglobin A1c (HbA1c) were assessed.
HN is a member of a class of novel mitochondrial-derived peptides released during mitochondrial dysfunction. HN reduces ROS production, enhances antioxidant protein expression, maintains redox balance, and suppresses TNF-α, IL-1β, and IL-6 to inhibit inflammation. Furthermore, resistance and endurance training has shown to increase HN expression in patients with prediabetes. Exercise – aerobic and endurance – has been shown to increase circulating and skeletal muscle levels of HN, correlating with improved insulin sensitivity and mitochondrial function.
Our results showed:
CT and AST supplementation both improved antioxidant defense and reduced inflammation, and their combination was more effective than either intervention alone.
CT and AST supplementation increased blood concentration of HN, and their combination showed greater effects than AST supplementation, but not CT.
CT and AST supplementation increased blood levels of miRNAs-126-3p, and -146a and decreased miRNA-122, with their combination being slightly more effective in decreasing miRNA-122.
Both interventions improved lipid profile, with their combination being more effective in improving HDL and TG levels, although not total cholesterol.
FBG, HOMA-IR, and HbA1c were reduced by CT but not by AST supplementation.
Our data suggest that combining exercise with AST supplementation might improve oxidative status and inflammation through mechanisms involving HN and miRNAs 122, 126-3p, and 146a. Alleviating OS and inflammation could, in turn, lead to improvements in lipid profiles (e.g., TG, and HDL), IR, and reductions in HbA1c and FBG, as observed in our study. Furthermore, the combined approach seems to be more effective at improving cholesterol and TG levels.“
A meta-analysis of randomized controlled trials reported until May 2025 assessed astaxanthin’s effects on lipid profiles. Neither of the two trials covered here nor the three trials covered in Part 1 were included in this meta-analysis.
“Astaxanthin, a xanthophyll carotenoid, has garnered significant interest due to its benefits with regard to dyslipidemia. This multifaceted functional food ingredient modulates several key enzymes associated with lipid regulation, including HMG-CoA reductase, CPT1, ACCβ, and acyl-CoA oxidase. It influences key antioxidant molecular pathways like Nrf2, limiting dyslipidemia occurrence and regulating liver cholesterol uptake through modulation of liver lipid receptors.
Astaxanthin daily doses and durations of analyzed studies: 12 mg for 8 weeks; 12 mg for 4 weeks; 20 mg for 12 weeks (two trials); 12 mg for 12 weeks; 8 mg for 8 weeks; 6 mg and 12 mg for 12 weeks; 6 mg, 12 mg, and 18 mg for 12 weeks.
This meta-analysis concludes positive effects of astaxanthin (6–20 mg/d) on HDL-C and triglyceride levels. Astaxanthin (6–20 mg/d) does not appear to significantly influence LDL-C and total cholesterol levels.
Regarding HDL-C, improvements were observed from 55 ± 8 mg/dL (pre-intervention) to 63 ± 8 mg/dL (post-intervention) (p < 0.01) in the 12 mg/d of astaxanthin groups. In triglyceride levels, results show a decrease from 151 ± 26 mg/dL (pre-intervention) to 112 ± 40 mg/dL (post-intervention) (p < 0.01) for 18 mg/d astaxanthin supplementation.
Further research is necessary to fully harness the potential of astaxanthin, which includes assessing astaxanthin in different subsets of patients, and in combination with other nutraceuticals to understand the compound’s effectiveness with regard to varying health conditions, genetic and epigenetic factors, and synergistic effects with other compounds.”
https://www.mdpi.com/1424-8247/18/8/1097 “Assessing the Effects of Moderate to High Dosage of Astaxanthin Supplementation on Lipid Profile Parameters—A Systematic Review and Meta-Analysis of Randomized Controlled Studies”
This same group of researchers assessed that in nine RCTs, astaxanthin had no effects on either body weight or BMI per https://www.mdpi.com/1424-8247/18/10/1482 “Therapeutic Potential of Astaxanthin for Body Weight Regulation: A Systematic Review and Meta-Analysis with Dose–Response Assessment”
Here are three 2025 clinical trials of the Nrf2 activator astaxanthin’s effects. Let’s start with a clinical trial of inflammation-related diabetic complications and insulin resistance:
“We investigated effects of 10 mg/day astaxanthin (ASX) supplementation for 12 weeks on microRNAs (miRNAs), lysophosphatidylcholine (LPC), and α-hydroxybutyrate (α-HB) as novel factors in development of a variety of diabetes-related complications.
LPC is believed to play a significant role in atherosclerosis and inflammatory diseases by modifying functions of multiple cell types, including smooth muscle cells, endothelial cells, monocytes, macrophages, and T cells. LPC can interfere with glucose-stimulated insulin secretion by impairing calcium homeostasis and other signaling pathways that are crucial for the proper functioning of beta cells. This impairment exacerbates hyperglycemia in diabetic patients. LPCs may impede insulin signaling pathways, thereby contributing to insulin resistance (IR).
α-HB is also an indicator of IR and impaired glucose regulation, both of which appear to result from excessive lipid oxidation and oxidative stress. The European population cohorts in 2016 identified α-HB as a selective biomarker for decreased glucose tolerance and prediabetes, which was independent of age, sex, BMI, and fasting glucose.
A number of studies have established a link between miR-21, miR-34a, and miR-155 and diabetic complications such as retinopathy and nephropathy.
In the ASX group, participants were divided into 2 subgroups according to the urinary albumin-to-creatinine ratio (ACR) (< 30 mg/g or ≥ 30 mg/g, an indicator of diabetic kidney disease).
The level of fasting plasma glucose before and after 12 weeks of treatment with ASX was 139.27 ± 21.18 vs. 126.43 ± 18.97 (p = 0.002), demonstrating a significant reduction compared to the placebo group.
In the ASX group, the mean HbA1c level at baseline was 7.89 ± 0.79 and declined to 7.05 ± 0.35 after the supplementation period, which was statistically significant.
Supplementation with ASX resulted in a statistically significant drop in HOMA-IR levels, whereas this parameter was not altered significantly in the placebo group.
The ASX group, in comparison with the placebo group, demonstrated marked changes in lipid profile factors such as TC, TG, and LDL (p = 0.011, p = 0.043, and p = 0.022, respectively).
Clinical studies indicate that rigorous diabetes management does not substantially diminish appearance of complications. Modifications in oxidative stress and IR markers, as well as miRNA expression, must be analyzed to identify biological markers with sufficient predictive power for development of complications in diabetic patients.
Supplementation with ASX substantially diminished the levels of α-HB, LPC, and inflammation-related miRNAs in diabetic patients with and without complications.”
Another clinical trial investigated astaxanthin’s effects in heart failure patients:
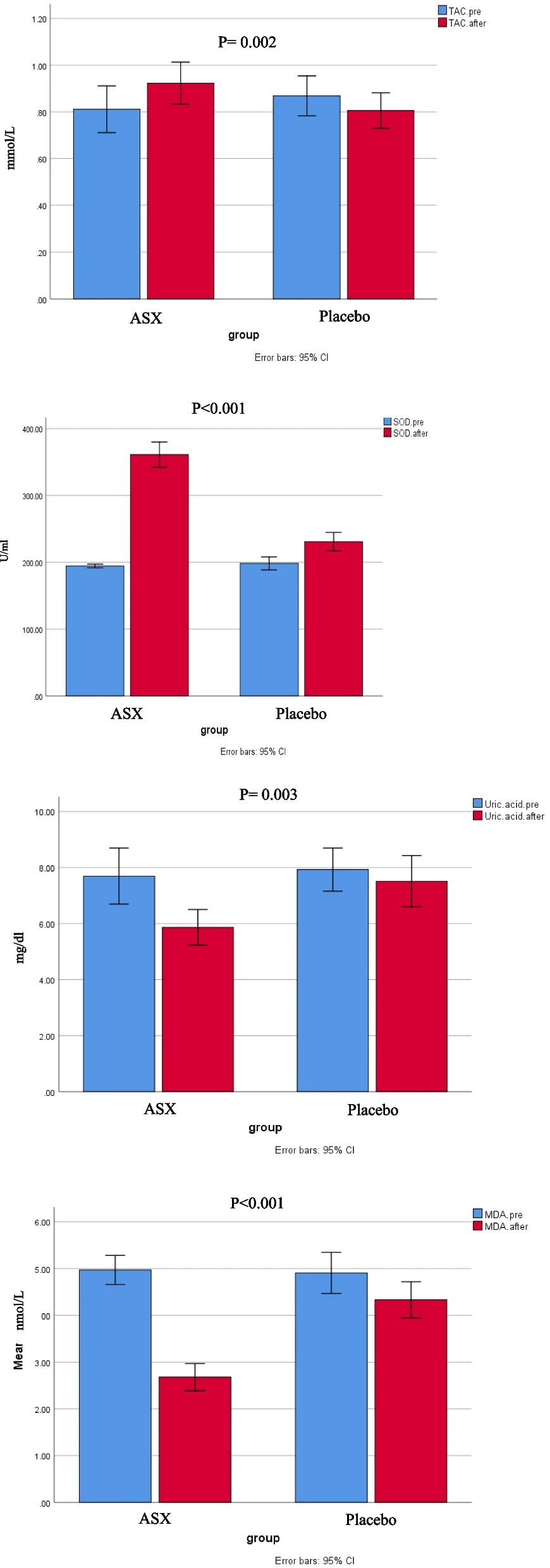
“Chronic heart failure (HF) is often linked to increased oxidative stress and metabolic issues like high uric acid, which can worsen outcomes.This study aimed to investigate the effects of ASX supplementation on oxidative stress markers as the primary outcome and clinical symptoms in patients with HF.
80 patients with HF were enrolled and randomly assigned to receive either ASX (20 mg/day) or a placebo (20 mg/day of maltodextrin) for 8 weeks. Biomarkers including total antioxidant capacity (TAC), malondialdehyde (MDA), superoxide dismutase (SOD), serum uric acid (UA), and clinical symptoms (dyspnea, fatigue, appetite) were assessed pre-and post-intervention.
Daily supplementation with 20 mg of ASX for eight weeks in patients with HF resulted in significantly greater improvements in oxidative stress biomarkers compared to placebo group. This improvement included reductions in uric acid and MDA, along increases in TAC and SOD.
In our study, participants received the cis-isomer form of ASX. The cis-isomer of ASX demonstrates greater anti-inflammatory and antioxidant properties than the trans-isomer, along with enhanced bioavailability. Inconsistencies among studies may be attributed to differences in participants’ baseline antioxidant status, underlying medical conditions, dosage, isomeric form and formulation of ASX used, and the duration of intervention.
One of the strengths of this study is that it represents the first randomized clinical trial to evaluate the effects of ASX supplementation on oxidative stress markers, UA levels, and clinical symptoms in patients with HF. Additionally, potential confounding factors were controlled as much as possible. However, several limitations were identified, including the relatively short intervention duration, limited sample size, limited generalizability of the findings due to the single-center design, absence of blood ASX level measurements, and lack of long-term follow-up.”
A third clinical trial evaluated astaxanthin’s effects as an adjunct to standard treatment of community-acquired pneumonia:
“Adult patients diagnosed with community-acquired pneumonia (CAP) were enrolled and assigned to receive either 12 mg/day ASX or a placebo in addition to standard antibiotic therapy for 7 days. Inflammatory markers, including interleukin-6 (IL-6), tumor necrosis factor-alpha (TNF-α), and interleukin-10 (IL-10), were measured at baseline and post-treatment. Secondary outcomes included Sequential Organ Failure Assessment (SOFA) and Acute Physiology and Chronic Health Evaluation II (APACHE II) scores.
A total of 80 patients (40 per group) completed the study. Patients receiving ASX exhibited significant reductions in pro-inflammatory cytokines compared to the placebo group. IL-6 and TNF-α levels were significantly lower in the ASX group at the end of the study (P < 0.05). Additionally, SOFA and APACHE II scores showed greater improvements in ASX-treated patients, suggesting a potential role in mitigating disease severity.
These findings suggest that ASX may help preserve organ function, limit the progression of inflammatory injury, and reduce overall disease severity in hospitalized patients with CAP.
ASX is widely regarded as the most potent carotenoid, owing to its unique molecular structure. Its polar-nonpolar-polar configuration enables it to span lipid bilayers and neutralize ROS both within and outside cellular membranes—an advantage not shared by other carotenoids that tend to localize at the membrane surface.
Despite the positive findings of this study, some limitations should nevertheless be considered.
The relatively small sample size may have limited the statistical power to detect differences in some outcomes and affects the generalizability of the findings.
Microbiological data on CAP pathogens were not collected. As different microorganisms can trigger distinct inflammatory responses, this limits our ability to assess pathogen-specific variations in ASX efficacy.
A notable limitation of this study is the short follow-up duration, with outcomes assessed only over a 7-day period. While this timeframe offers insight into the acute effects of ASX on inflammatory and OS markers, it does not clarify whether these benefits are sustained beyond the immediate treatment window.
The fixed dose of 12 mg once daily may not have maintained optimal therapeutic levels throughout the day. Dose-ranging studies and evaluations of alternative regimens are needed to determine the most effective strategy.”
Wrapping up Plasmalogens Week with a summary of my plasmalogen-related experiences over the past two years since Plasmalogens, Part 3 in November 2023.
I took detailed plasmalogen measurements on July 24, 2025, with Dr. Goodenowe’s BioScan product. I’d guess that the populations against which BioScan personal Z-scores are derived are from Dr. Goodenowe’s research during this century, many frozen samples of which he’s kept. If so, I’d guess that these populations’ data probably don’t have bell-shaped curves, and that their data’s means and standard deviations may be skewed as they’re representing people who were diseased and/or old.
Here’s my peroxisomal function panel:
I wasn’t taking ProdromeNeuro or ProdromeGlia at the BioScan blood draw time. ProdromeNeuro and ProdromeGlia supplements contain plasmalogen precursors that bypass peroxisome organelles’ normal plasmalogen synthesis functions. I haven’t reordered these supplements in 2025, but took them until my supplies ran out in January 2025. Don’t know to what extent their effects may have continued for six months.
Every day for months before the BioScan, I took a fish oil capsule with 690 mg EPA and 310 mg DHA, and a flax seed oil capsule (700 mg alpha linolenic acid omega-3, 154 mg linoleic acid omega-6, and 168 mg oleic acid omega-9). I also ate 3 eggs a day.
These practices influenced the above peroxisomal function results. My Z-scores of DHA and EPA ethanolamine plasmalogens (DHA +1.3, EPA +1.7) are more than one standard deviation above their respective population means.
The next step of plasmalogen synthesis after peroxisomes takes place in endoplasmic reticulum organelles. Among other papers describing these steps in the ER link’s results, Improving peroxisomal function states:
“Proper functioning of peroxisomes in metabolism requires the concerted interaction with other subcellular organelles, including the endoplasmic reticulum (ER), mitochondria, lipid droplets, lysosomes, and the cytosol. A striking example of peroxisome-ER metabolic cooperation is de novo biosynthesis of ether phospholipids.”
ER stress involves the unfolded protein response, a protein homeostasis-maintaining system that monitors ER conditions by sensing inadequacy in ER protein folding capacity. ER stress is a very common occurrence for humans, in part because ER protein folding has an over 80% failure rate per Every hand’s a winner, and every hand’s a loser.
I haven’t read papers about ER stress directly influencing plasmalogen abundance. But I’ve curated papers, including several during this Plasmalogens Week, that demonstrate how oxidative stress reduces plasmalogens.
Here’s my BioScan inflammation / oxidative stress panel:
Back to my peroxisomal function panel: I don’t consider my negative Z-scores (below the population mean) of Total PEs and Total PCs to be actionable. Both of them produced positive Z-scores (above the population mean) of their respective total plasmalogens (Total PLEs +1.3, Total PLCs +0.5). I view Total PEs and Total PCs as pools of raw materials for plasmalogen synthesis that are used when needed.
My July 2025 BioScan shows that my current practices provide adequate plasmalogens as compared with unknown populations. It indicates that to produce adequate plasmalogens, I don’t need ProdromeNeuro and ProdromeGlia plasmalogen precursor supplements that bypass normal peroxisomal function plasmalogen synthesis.
This year’s BioScan was a one-time event. I don’t agree with advocates for constantly tweaking health parameters, or obtaining frequent test results for ‘youthful’ targets, or competing with or conforming to other people’s measurements, or unfounded longevity beliefs. It’s every human’s choice whether or not we take responsibility for our own one precious life. Being overly obsessed about one’s health can be among the many symptoms of what’s ruining a person’s life.
I might use a future version of BioScan along with ProdromeNeuro and ProdromeGlia plasmalogen precursor supplements if I had to recover from an accident or some other health emergency that creates a substantial demand for plasmalogens’ antioxidant activities. But I’d first return to past practices I’ve found to be successful in combating oxidative stress, like increasing the frequency of Nrf2 activation by eating broccoli sprouts twice a day rather than once daily.
Continuing Plasmalogens Week with three 2025 papers, starting with a rodent study of genetically deleting a plasmalogen catabolizing enzyme:
“In this study, we investigated the impact of global and tissue-specific loss-of-function of a plasmalogen catabolizing enzyme, lysoplasmalogenase (TMEM86B), on circulatory and tissue lipidomes. Mice with homozygous global inactivation of Tmem86b (Tmem86b KO mice) were viable and did not display any marked phenotypic abnormalities.
Tmem86b KO mice demonstrated significantly elevated levels of plasmalogens alkenyl phosphatidylethanolamine (PE(P)) and alkenyl phosphatidylcholine (PC(P)), as well as lysoplasmalogens, in the plasma, liver, and natural killer cells compared to their wild-type counterparts. The endogenous alkenyl chain composition of plasmalogens remained unaltered in Tmem86b KO mice. Consistent with the global knockout findings, hepatocyte-specific Tmem86b knockout mice also exhibited increased plasmalogen levels in the plasma and liver compared to their floxed control counterparts.
Plasmalogens may be synthesized locally within various tissues, with each organ possessing the necessary enzymatic machinery to regulate its own plasmalogen levels. Plasmalogens are important structural constituents of the biological membranes of animals and certain anaerobic bacteria, and have several well-described functions, including regulating membrane dynamics and vesicular cholesterol transport and homeostasis.
One of the most interesting features of plasmalogens is their endogenous antioxidant activity, which is mostly due to the vinyl ether bond, which can scavenge reactive oxygen species and thereby protect other biomolecules from oxidative damage.
They increase the gene expression of multiple antioxidant enzymes to protect against chemically induced cytotoxicity and lipid peroxidation in cultured hepatocytes.
Plasmalogen derivatives such as polyunsaturated fatty acids (AA or DHA) and lysoplasmalogens can act as lipid mediators for multiple cellular signaling activities.
Plasmalogens are important for phagocytosis of macrophages, lipid droplet formation, and development and function of neuromuscular junctions.
They play vital roles in mediating immune responses, and mitochondrial fission to regulate adipose tissue thermogenesis, and protecting neuronal cells against cell death and inflammation.
All of these are suggestive of a critical role played by plasmalogens in maintaining cellular homeostasis.
While plasmalogen anabolism is well defined, its catabolism has been less studied. During catabolism, plasmalogens are deacylated by the action of a calcium-independent phospholipase A2 enzyme (iPLA2) to produce lysoplasmalogens. However, cytochrome C has also been shown to act as a plasmalogenase under certain circumstances.
The amount of lysoplasmalogens in cells is tightly regulated either by reacylation into plasmalogens through a coenzyme A-independent transacylase, or by degradation into fatty aldehydes and glycerophospholipids by an alkenyl ether hydrolase commonly known as lysoplasmalogenase. Lysoplasmalogenase is a microsomal transmembrane enzyme highly specific for lysoplasmalogens, and has no activity against plasmalogens.
While research on the distinct biological functions of lysoplasmalogens and plasmalogens is lacking, some reports indicate potential toxic effects of lysoplasmalogens. Degradation products of lysoplasmalogens, such as fatty aldehydes, are highly reactive electrophilic compounds that can form toxic adducts with cellular proteins and lipids. These interactions can lead to cellular dysfunction and contribute to various pathological conditions. Their accumulation in ischemic/reperfused tissues has been associated with cellular damage.
However, we observed that the amount of lysoplasmalogens as a proportion of total plasmalogens in the liver of Tmem86b KO mice was only ∼3.5%, indicating that elevated lysoplasmalogens are rapidly converted into plasmalogens within the liver. In adipose tissue-specific Tmem86a KO mice, which also exhibited higher lysoplasmalogens, no toxic effects were observed. Instead, these mice showed elevated mitochondrial oxidative metabolism and energy expenditure, offering protection from high-fat diet-induced metabolic dysfunction. These findings suggest that any potential toxic effects of lysoplasmalogens are largely mitigated by their rapid reacylation into plasmalogens.
This study enhances our understanding of regulatory mechanisms governing plasmalogen metabolism, and highlights the potential of targeting Tmem86b to therapeutically raise plasmalogen levels.”
An independent researcher published a commentary on the above study:
“While the biosynthesis of this particular lipid subclass, starting in the peroxisomes and ending at the endoplasmic reticulum, has been the subject of extensive research, the degradation pathway of these compounds remains to be further elucidated. Plasmalogen breakdown is a complex process involving enzymatic hydrolysis, oxidative cleavage, and possibly also a recycling mechanism.
A fundamental unresolved question in the field of plasmalogen catabolism is which of the two possible reaction routes is actually the more important one. Either 1) directly via plasmalogenase or 2) via a deacylation step by a plasmalogen-specific phospholipase A2 (cPLA2, PLA2G4A), yielding a lysoplasmalogen as the first degradation product, and subsequent hydrolysis of the ether bond by a lysoplasmalogenase such as TMEM86A and TMEM86B. It is also unclear how these pathways interact or compensate for each other, how they are regulated, and whether they are tissue- or cell type–specific.
To make the story even more complex, a CoA-independent transacylase activity was described that reacylates lysoplasmalogen intermediates back to plasmalogens by transferring polyunsaturated fatty acids to the vacant sn-2 position of ether lysophospholipids. But no gene for this enzyme has so far been identified.
Why is plasmalogen breakdown so important? Disturbances in plasmalogen metabolism are associated with several human disorders. Neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease, and multiple sclerosis have been shown to be associated with reduced levels of plasmalogens.
Unfortunately, it is still too early to draw conclusions about the individual roles of TMEM86A and TMEM86B, as their cellular localisation and function are not sufficiently studied, and reliable antibodies for these proteins are not yet available. Localization of the two TMEM86 homologs overlaps to some extent, as shown, for example, by their gene expression in small intestine. However, whether one isoform is able to compensate for a deficiency in the other is uncertain, and was not found in small intestine of Tmem86b knockout mice [in the above study].
In contrast to the two proteins TMEM86A and TMEM86B, cytochrome c is much better studied. It is associated with the inner mitochondrial membrane, and can be released into the cytosol during apoptosis. It has a wide tissue distribution with most abundant gene expression levels in the digestive tract and heart.“
The statement “no gene for this enzyme has so far been identified” revealed a paradigm. But maybe what’s being observed evolved before genes?
One example of this principle is from the 1966 https://www.science.org/doi/10.1126/science.152.3720.363 “Evolution of the Structure of Ferredoxin Based on Living Relics of Primitive Amino Acid Sequences” which provided evidence pointing to heme protein evolution beginning before gene evolution. Its abstract included this statement:
“We explain the persistence of living relics of this primordial structure by invoking a conservative principle in evolutionary biochemistry: The processes of natural selection severely inhibit any change in a well-adapted system on which several other essential components depend.”
Maybe the process of reassembling plasmalogen breakdown products back into plasmalogens without involving a specific gene likewise became essential?
A role of plasmalogens in diabetic kidney disease was found in a third study that investigated a genetic rodent model of diabetes:
“Diabetic nephropathy (DN) represents a frequent cardiovascular complication of diabetes, affecting about 20–50% of individuals with the disease. Globally, it constitutes a primary etiology for end-stage kidney disease (ESKD) and chronic kidney disease (CKD), while also serving as a significant independent risk factor for cardiovascular morbidity and mortality.
Although intensive management strategies targeting blood pressure and glucose levels demonstrably attenuate the risk of DN development, they do not confer complete protection. This residual risk strongly implicates pathogenic factors beyond impaired glucose metabolism and hemodynamic alterations in DN pathogenesis.
In the present study, we employed the db/db mice as the DN model. When compared to other diabetes models, such as those induced by streptozotocin (STZ) or high-fat diet combined with STZ, the db/db model more accurately recapitulates the pathological features of human type 2 diabetes mellitus (T2DM). It also possesses a stable genetic background, making it particularly well-suited for the investigation of diabetes complications.
Transcriptomics revealed extensive dysregulation of metabolic and lipid regulatory pathways in db/db. Lipidomics uncovered pronounced abnormalities in cardiolipin species composition and plasmalogen profiles. Transcriptome-lipidome integration demonstrated impaired phosphatidylcholine (PC) biosynthesis, mechanistically linked to dysregulation of choline phosphotransferase 1 (chpt1), which correlated significantly with compromised tissue regeneration capacity.
Volcano plot analysis delineated specific lipid alterations, particularly in plasmalogen species in plasmalogen lipids. Plasmenylcholines (plas-PC) and plasmenylethanolamine (plas-PE) containing n-3 polyunsaturated fatty acids (PUFAs) were significantly decreased in the kidneys of db/db mice. Conversely, plas-PCs and plas-PEs esterified with n-6 PUFAs showed substantial accumulation in diabetic kidneys.
In conclusion, the highly sensitive and extensively targeted UHPLC-MS/MS methodology enabled a more in-depth characterization of renal metabolic and lipid perturbations in db/db mice. These alterations principally reflect the sustained inflammatory milieu and compromised antioxidant defenses characteristic of DN renal tissues.”
Continuing Part 1 with three 2025 papers, starting with a rodent study of dietary mussel plasmalogens’ effects on atherosclerosis:
“The purpose of this study was to clarify the underlying mechanisms of Mytilus edulis-derived plasmalogens (Pls) against atherosclerosis (AS) in ApoE−/− mice induced by a high-fat diet (HFD), through a comprehensive analysis of hepatic metabolomics and aortic transcriptomics data. Besides Pls role as the storage pool of n-3 PUFAs, the structural feature of vinyl ether bond at sn-1 position confers multiple advantages upon Pls compared to their diacyl counterparts, including enhanced antioxidant capacity, increased membrane fluidity, as well as improved stability and stability of biomembranes.
The C57BL/6 mouse strain is susceptible to high-fat diet (HFD)-induced AS lesions, and ApoE knockout accelerates AS development. Molecular mechanisms by which Pls ameliorate AS were investigated through a comprehensive analysis of hepatic metabolomics and aortic transcriptome profiles, focusing on changes in gene related to the p38 mitogen-activated protein kinase (MAPK) signaling pathway and the downstream inflammatory response.
The concentration of Pls in mussel tissues is 32 μgmg−1 (dry weight), and the obtained Pls contains 49.53% of phosphatidylethanolamine-Pls, 35.87% of phosphatidylcholine-Pls, and 14.60% of phosphatidylserine-Pls. The main fatty acid compositions of Pls are presented in Supplementary Table 1, which indicates that EPA accounts for 45.82% and the n-3/n-6 ratio is 3.84.
Pls inhibited aortic lipid accumulation, prevented thickening of the aortic wall, and suppressed collagen accumulation at the aortic-heart junction. Pls inhibited HFD-induced loosening of hepatocyte arrangement, vacuolization, and accumulation of lipid droplets.
Although several key components of MAPK signaling pathway were suppressed at both the transcriptional and protein levels in Pls-treated mice, no significant changes in phosphorylated p38 protein were observed among the experimental groups in our study. Further research is needed to elucidate the overall inhibitory mechanism of Pls on p38 protein and the MAPK signaling pathway.”
A rodent / human cell study investigated effects of plasmalogens in innate immune system macrophages on atherosclerosis:
“We demonstrate that simultaneous inactivation of two key enzymes involved in macrophage polyunsaturated fatty acid (PUFA) metabolism—ELOVL5, which elongates long-chain PUFAs, and LPCAT3, which incorporates them into phospholipids—disrupts membrane organization by promoting the formation of cholesterol-enriched domains. This increases macrophage sensitivity to cytotoxic oxysterols and leads to more vulnerable atherosclerotic plaques with enlarged necrotic cores in a mouse model of atherosclerosis.
We identified ELOVL5 as one elongase facilitating the conversion of C20 to C22 PUFA. In humans, analysis of 187 carotid plaques reveals a positive correlation between LPCAT3/ELOVL5-generated phospholipids—including arachidonate (C20:4 n-6)-containing ether lipids—and more stable plaque profiles. Additionally, Mendelian randomization analysis supports a causal relationship between LPCAT3 expression and reduced risk of ischemic stroke.
Potentially beneficial effects we observed in mice and in human atheroma plaques were mainly associated with PLs enriched in omega-6, particularly in AA. Although omega-6 FAs are often considered as pro-inflammatory, their role is undergoing reconsideration, with markers linked to the intake of omega-6 appearing beneficial in the context of cardiovascular diseases. In this context, it is worth to note that AA-containing plasmalogens have been previously identified as markers of healthy obesity.
Our findings uncover a regulatory circuit essential for PUFA-containing phospholipid generation in macrophages, positioning PUFA-containing ether lipids as promising biomarkers and therapeutic targets.”
A human study included plasmalogens in investigating associations among people with mental illness and their lipid profiles:
“Plasma lipidomic profiles of 623 individuals (188 schizophrenia (SCZ), 243 bipolar disorder (BD), 192 healthy controls) belonging to the PsyCourse Study were assessed using liquid chromatography and untargeted mass spectrometry. Exact etiology of these major mental health disorders is yet unknown and while their symptoms overlap, their diagnostic criteria are based on clinical evaluations of symptoms without objective markers.
Cognitive dysfunction is among the most disabling symptoms of SCZ and BD, and is difficult to treat with the commonly used pharmacologic regimes. Consequently, it has important impacts on long-term functional outcomes.
We aimed to answer the question, whether specific lipid species or classes were associated with differential performance across various cognitive domains, including psychomotor and processing speed, executive function, short-term and working memory and crystalized intelligence and whether these associations were affected by diagnoses.
Lipids belonging to the phosphatidylethanolamine plasmalogen (PE-P) class emerged as the main lipid class associated negatively with DG-SYM test performance, representative of processing and psychomotor speed. Our findings showed that higher levels of PE-P 42:5, PE-P 40:4, PE-P 40:5, and ceramide 38:1 in plasma samples of our study are significantly associated with poorer DG-SYM test performance. The DG-SYM test mainly measures processing speed, the amount of time required to complete a series of cognitive tasks. Enrichment analysis also showed significant associations between other lipid classes and various cognitive tests.
Our findings suggest a link between lipids and cognitive performance independent of mental health disorders. Independent replication is warranted to better understand if phosphatidylethanolamines could represent an actionable pharmacologic target to tackle cognitive dysfunction, an important unmet clinical need that affects long-term functional outcomes in individuals with severe mental health disorders.”
It was apparently beyond these researchers’ expertise to offer informed discussion on this study’s associative link between enrichment of these three phosphatidyl ethanolamine plasmalogens and cognitive dysfunction. Grok countered that their depletion was associated with neurodegenerative diseases (Alzheimer’s, Parkinson’s, multiple sclerosis), cardiovascular risk / oxidized-LDL burden, and chronic fatigue / post-viral syndromes.
Continuing Plasmalogens Week with three 2025 papers, starting with a human study that included plasmalogen biomarkers of non-communicable disease fatigue symptoms:
“This study explored the biological mechanisms underlying fatigue in patients with NCDs using a multi-omics approach. Our findings indicate that distinct metabolic pathways, salivary microbiota, and genetic factors may contribute to different dimensions of fatigue, including general, physical, and mental fatigue.
General fatigue is associated with unsaturated fatty acid biosynthesis, indicating its role in lipid metabolism.
Physical fatigue was associated with plasmalogen synthesis, mitochondrial beta-oxidation of long-chain fatty acids, and selenoamino acid metabolism, suggesting a potential contribution of impaired energy production.
Mental fatigue is associated with homocysteine degradation and catecholamine biosynthesis, which may influence cognitive fatigue.
This exploratory study suggests that fatigue in patients with NCDs may involve disruptions in lipid metabolism, neurotransmitter pathways, microbial composition, and genetic variations. Blood-based biomarkers showed better predictive potential for physical fatigue, whereas salivary-based models were more indicative of mental fatigue.
Although our findings support the role of lipid metabolism, the contribution of plasmalogen synthesis remains underexplored. Further studies are needed to validate these findings and understand their mechanisms of action.”
A human study of metabolic dysfunction-associated steatotic liver disease (MASLD) included investigating plasmalogens:
“In this study, we applied untargeted metabolomic profiling to serum samples from individuals with and without MASLD, classified by the Fatty Liver Index, with the goal of identifying characteristic metabolic signatures and pathways that may underlie disease presence and progression. Individuals in the MASLD group displayed significantly higher levels of ALT, AST, ALP, and GGT, reflecting ongoing hepatic injury, cholestasis, and oxidative stress. However, albumin and bilirubin levels remained within normal limits, indicating early to intermediate disease stages rather than advanced fibrosis or cirrhosis.
A consistent and highly significant lipidomic pattern in the MASLD group is the depletion of plasmalogens and sphingomyelins. Depletion of these lipid classes was identified as a hallmark of insulin resistance as defined by the triglyceride-glucose index. In contrast, phosphatidylcholine, phosphatidylethanolamine, and phosphatidylinositol species were elevated in MASLD, pointing toward broader lipid remodeling events.
Reduced plasmalogen and sphingomyelin levels positions their depletion as a core feature of metabolic dysfunction. Plasmalogens are ether phospholipids with strong antioxidant capacity, and their reduction suggests a loss of protective buffering against oxidative stress, one of the main drivers of MASLD progression. Similarly, sphingomyelin depletion implicates altered membrane dynamics and signaling disturbances, further contributing to metabolic dysfunction.
Depletion of plasmalogens 1-(1-enyl-palmitoyl)-2-oleoyl-GPC (P-16:0/18:1), 1-(1-enyl-palmitoyl)-2-linoleoyl-GPC (P-16:0/18:2), 1-(1-enyl-palmitoyl)-2-palmitoyl-GPC (P-16:0/16:0), 1-(1-enyl-palmitoyl)-2-palmitoleoyl-GPC (P-16:0/16:1), 1-(1-enyl-palmitoyl)-2-oleoyl-GPE (P-16:0/18:1), 1-(1-enyl-palmitoyl)-2-linoleoyl-GPE (P-16:0/18:2), and disruption of the glutamate–gamma-glutamyl pathway stand out as central features of metabolic dysfunction in MASLD, with clear potential to inform biomarker discovery, disease classification, and the design of targeted therapeutic strategies.”
https://www.mdpi.com/2218-1989/15/11/687 “Metabolomic Signatures of MASLD Identified by the Fatty Liver Index Reveal Gamma-Glutamyl Cycle Disruption and Lipid Remodeling”
A rodent study investigated dietary sea squirt (AM) plasmalogen ethanolamine (PlsEtn) extract’s and dietary pig liver (PL) phosphatidyl ethanolamine (PtdEtn) extract’s effects on acetaminophen liver injury:
“We investigated dietary effects of PlsEtn from ascidian on chronic hepatic injury in acetaminophen (APAP)-treated mice. Five-week-old male mice were divided into four groups (n = 12), which were treated with experimental diets for two weeks and then the respective APAP-containing diet for five weeks.
Ingested PlsEtn is digested into lysoPlsEtn and free fatty acid in the small intestine. PlsEtn digests are absorbed and are subsequently resynthesized into PlsEtn preferentially with PUFA.
Acetaminophen is a frequently used analgesic and antipyretic. Approximately 90% of APAP is metabolized by UDP-glucuronosyltransferase and sulfotransferase into glucuronic acid and sulfate conjugates, respectively.
5–9% of APAP is metabolized into the highly reactive intermediate N-acetyl-p-benzoquinone imine (NAPQI). This metabolite is considered a pivotal molecule in APAP-induced hepatotoxicity and is conjugated by glutathione (GSH). Excessive NAPQI levels deplete GSH and covalently bind to cellular proteins, resulting in organelle dysfunction, such as mitochondria dysfunction. These impairments induce oxidative stress, cell malfunctions, and subsequently, cell death, such as ferroptosis and apoptosis.
Mice were treated with continuous APAP consumption to induce oxidative stress and impaired lipid metabolism in the liver. Effects of diets were evaluated based on levels of malondialdehyde (MDA), a marker of lipid oxidation, on fatty acid content, and on expression of apoptosis-related proteins in the liver.
The PlsEtn-rich diet effectively suppressed APAP-induced decrease in body and liver weights of mice. However, this suppressive effect was not observed in mice fed a PtdEtn-rich diet. APAP administration decreased the total fatty acid content in the liver, whereas a PlsEtn-rich diet alleviated this decrease and increased the hepatic content of docosahexaenoic acid (DHA).
Owing to the alkenyl linkage, which exhibits antioxidant properties, PlsEtn was expected to markedly suppress hepatic lipid oxidation. However, its suppressive effect was the same extent as that by PtdEtn. Both PlsEtn and PtdEtn contain an ethanolamine base in their structures, and free ethanolamine and its metabolite choline suppress lipid peroxidation. Dietary PlsEtn and PtdEtn may be metabolized into free ethanolamine and its further metabolites, which may alleviate APAP-induced hepatic lipid oxidation.
Dietary ethanolamine glycerophospholipids (EtnGpls) rich in PlsEtn or PtdEtn suppressed APAP-induced lipid oxidation in the liver. Protein expression results revealed that dietary EtnGpls reduced expression of certain apoptosis-related proteins compared to the APAP group. This reduction was more effective in mice fed the PlsEtn-rich diet than in those on the PtdEtn-rich diet.”
https://www.mdpi.com/2076-3417/15/11/5968 “Dietary Ethanolamine Plasmalogen from Ascidian Alleviates Chronic Hepatic Injury in Mice Treated with Continuous Acetaminophen”
This study neither demonstrated nor provided citations for its dietary plasmalogen recycling statements.
Three more plasmalogen health and disease papers are curated in Part 2.