A 2026 primate study investigated effects of vitamin C:
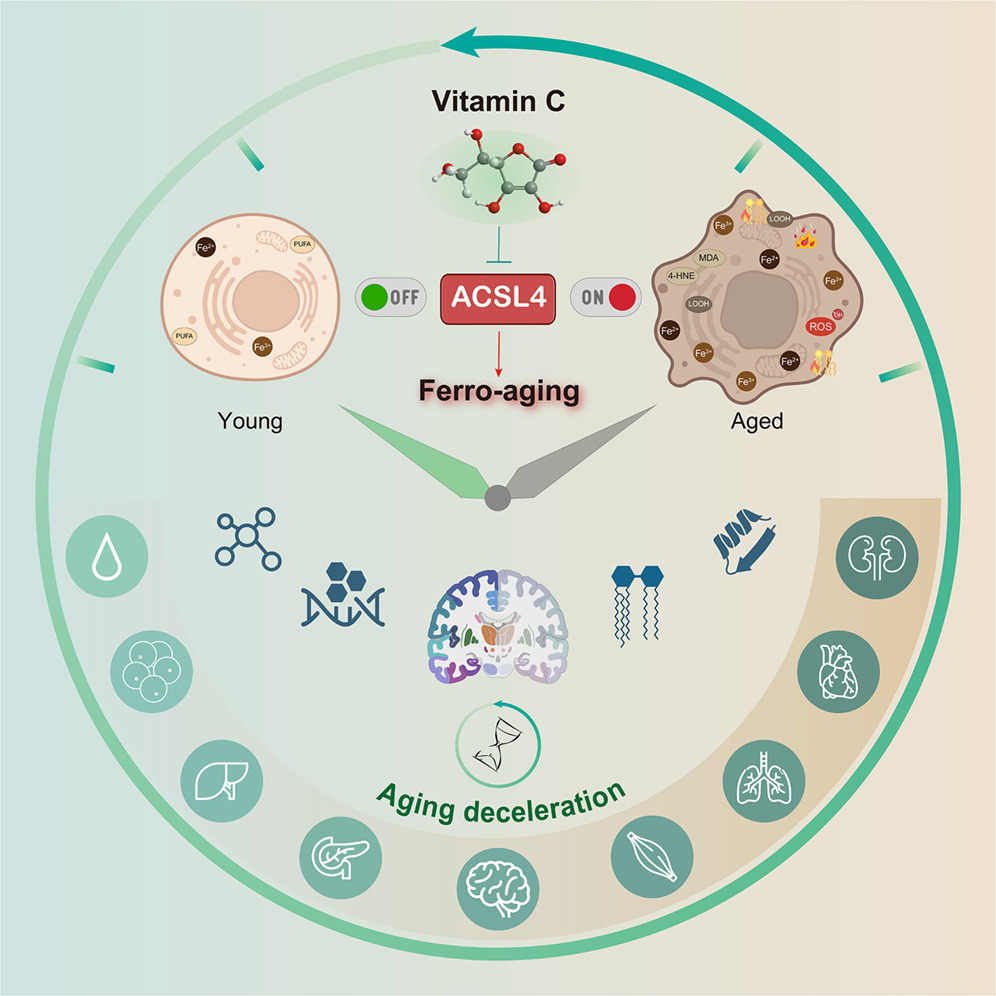
“Here, we define a conserved iron-lipid axis driving primate aging, termed ‘ferro-aging.’ Multi-tissue profiling in humans and non-human primates reveals age-progressive iron accumulation, fueling chronic lipid peroxidation orchestrated by acyl-coenzyme A (CoA) synthetase long-chain family member 4 (ACSL 4). Distinct from acute ferroptosis, this ACSL4-mediated process promotes cellular senescence and systemic functional decline.
We identify vitamin C (VC) as a direct inhibitor of ACSL4. Long-term VC administration in aged monkeys for over 40 months potently reduces ferro-aging signatures across tissues, attenuates multi-organ pathology, and improves neurological and metabolic functions. Multi-omic aging clocks indicate the VC-mediated reversal of biological age.
Despite decades of interest in oxidative stress, largely sparked by the free radical theory of aging, efforts to modulate it broadly with antioxidants have yielded inconsistent or neutral outcomes, highlighting the theory’s limitations and underscoring the need to identify more specific, upstream drivers. A critical challenge remains: determining whether the iron-lipid axis constitutes a core upstream driver of aging in primates and, if so, whether it is therapeutically targetable.
In this study, we bridge these gaps. We define an iron-triggered, ACSL4-governed, lipid peroxidation-driven program that escalates with age across diverse cell types and multiple organs in non-human primates.
VC treatment dose-dependently increased Nrf2 phosphorylation and activation. VC orchestrates a dual-defense strategy against ferro-aging: it directly suppresses the pro-aging lipid peroxidation driver ACSL4, while in parallel, it bolsters the cell’s intrinsic antioxidant capacity via Nrf2 pathway activation.
Middle-aged cynomolgus monkeys (12–16 years old, approximating human 40–50 years) received daily oral VC (30 mg/kg group) or a control treatment for 40 months under standardized conditions.
Structural MRI analysis demonstrated that VC intervention counteracted age-related brain atrophy. Using general linear mixed models, we found that VC restored cortical surface area in the frontal lobes of aged monkeys. Regional analysis identified enlargement in four regions of the orbital frontal cortex, an area critical for adaptive behavior.
Diffusion MRI-based connectomics revealed that, compared with young animals, aged monkeys exhibited reduced structural connectivity in 18 brain regions. VC treatment restored connectivity in 9 of these regions, which were predominantly located in the posterior parietal cortex, a hub for spatial awareness and decision-making.
VC exerted robust neuroprotective effects. It attenuated heterochromatin loss (increased H3K9me3) in the prefrontal cortex and hippocampus and reduced abnormal protein aggregates, including cytosolic aggresomes and Aβ. Additionally, VC lowered the abundance of activated microglia and astrocytes and suppressed expression of the innate immune sensor cGAS in the hippocampus.
VC supplementation reduced the estimated biological age across multiple organs. At the epigenetic level, VC lowered DNA methylation age in several tissues, including brain, brown adipose tissue, muscle, skin, aorta, and kidney. In the hippocampus, the most substantial reductions in biological age occurred in microglia, oligodendroglia, and oligodendrocyte precursor cells. In the pancreas, alpha cells, beta cells, and ductal cells showed the greatest rejuvenation.
In summary, chronic VC supplementation inhibits the ferro-aging pathway, reduces multidimensional biological age across primate organs, and ameliorates a spectrum of functional declines in nervous and metabolic systems. Our work establishes ACSL4 inhibition as a promising and translationally relevant therapeutic strategy for mitigating aging-related decline.
A long-term, 40-month intervention study in aged non-human primates is a highly translational model given their shared inability with humans to synthesize VC endogenously. The finding that a single, safe nutrient can reverse multidimensional aging clocks in a primate has profound implications for translational longevity medicine.”
https://www.sciencedirect.com/science/article/abs/pii/S1550413126000537 “Vitamin C inhibits ACSL4 to alleviate ferro-aging in primates” (not freely available) Thanks to Dr. Pradeep Reddy for providing a copy.
Grok’s take on this study:
“For humans (who, like macaques, cannot synthesize vitamin C), the Recommended Dietary Allowance (RDA) is 75–90 mg/day for adults (~1–1.5 mg/kg for a 60–70 kg person) to prevent deficiency. Upper safe intake levels are much higher: up to 2,000 mg/day (Tolerable Upper Intake Level) is considered safe for most adults, with no established adverse effects at that level from food/supplements.
Treated monkeys represent advanced aging stages (likely equivalent to human 50s–70s+ based on ‘aged’ designation and long-term intervention effects), extending the prior 12–16-year monkey range (human ~35–55) to broader anti-aging applications. While human trials are needed, the primate evidence (long-duration, systemic benefits) strengthens the case for high-dose, sustained vitamin C as a strategy against ferro-aging in humans. It elevates vitamin C from a nutrient to a targeted anti-aging compound in primates.”
Coincidentally, I started taking extra vitamin C separately from other supplements in the form of liposomal 1 gram twice daily this past winter. Can’t say that it had any effects on my intended target, avoiding sniffles and sneezing, as allergy season kicked off in early February. With this study’s findings, I’ll continue.

























