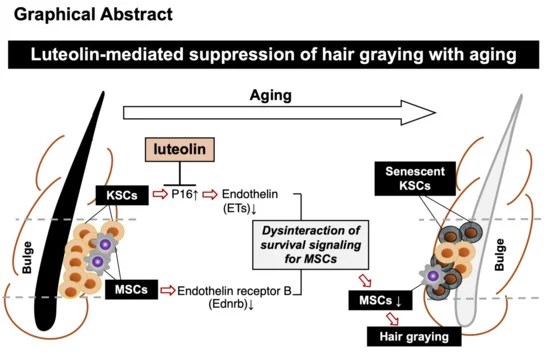
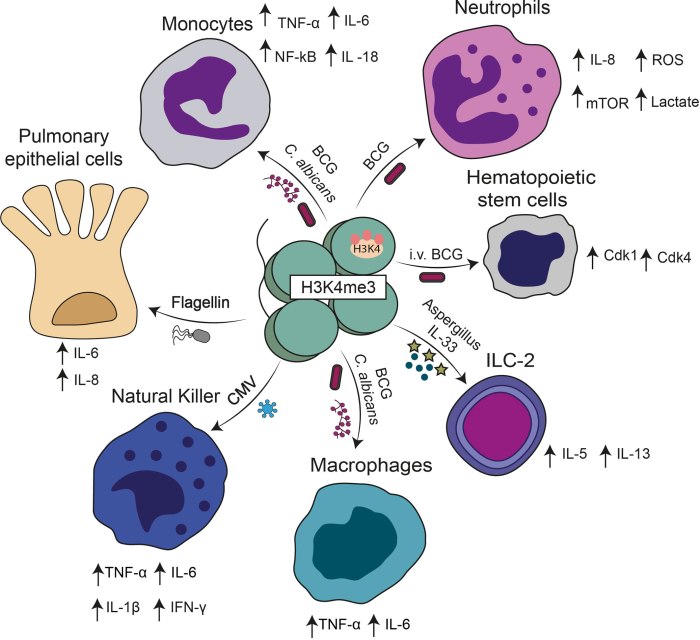
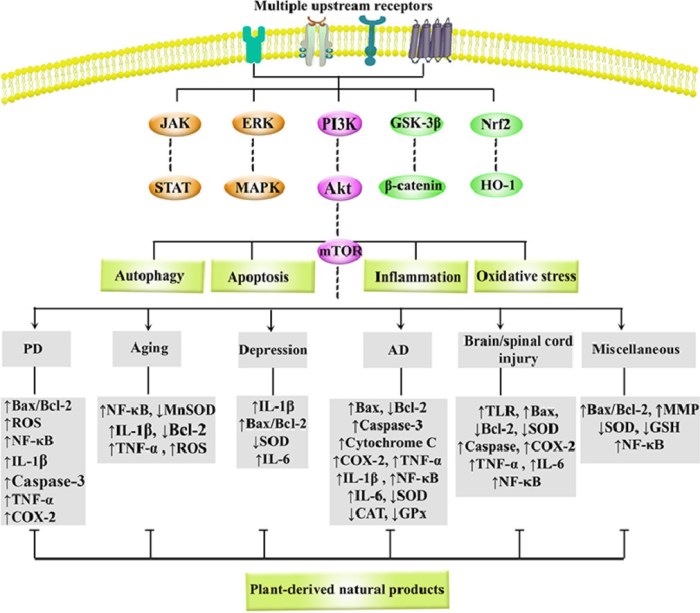
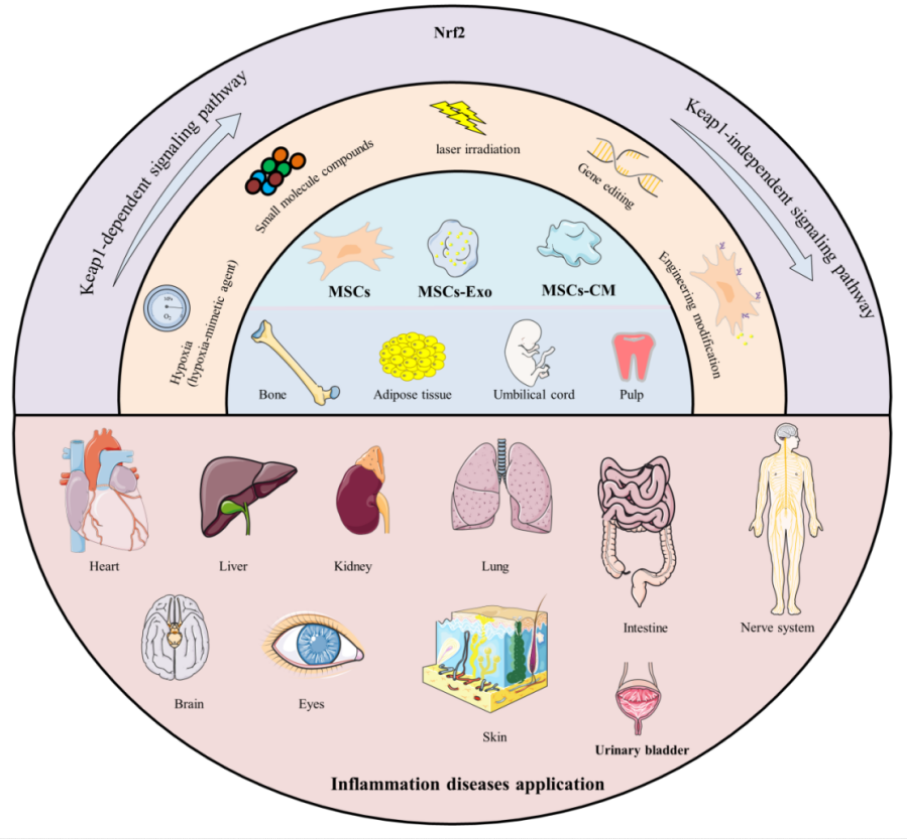
A 2026 review subject was mechanisms and therapeutic potential for Nrf2 activators in combination with mesenchymal stem cells:
“Mesenchymal stromal/stem cells (MSCs) are multipotent stem cells that can be isolated from various tissues – such as bone marrow (BM), umbilical cord (UC), adipose tissue (AD), dental pulp (DP), hair follicle (HF), and placenta – and differentiated into multiple lineages under appropriate conditions. Their functional repertoire includes immunomodulation, homing, and differentiation, which collectively help establish a balanced inflammatory and regenerative niche within damaged tissues during severe inflammation. MSCs-derived extracellular vesicles (MSCs-EVs) and conditioned medium (MSCs-CM) play remarkable roles, exhibiting potent anti-inflammatory and antioxidant properties that offer novel therapeutic alternatives for inflammatory diseases.
Therapeutic capacity of MSCs in inflammatory conditions is increasingly attributed to their potent paracrine activity rather than solely to their differentiation potential. A key mechanism underlying this paracrine effect is activation of the Nrf2 antioxidant pathway.
MSCs and their secreted products including exosomes (Exos), EVs, and CM, activate Nrf2 through multi-dimensional/target mechanisms, thereby enhancing cellular antioxidant defenses, modulating immune responses, and promoting tissue repair. It is noteworthy that therapeutic efficacy of MSCs and their derivatives can be enhanced through external modulation, including pretreatment with natural compounds.
Preconditioning refers to brief treatment of MSCs or their derivatives with physical, chemical, or biological factors prior to application, aiming to enhance their ability to counteract oxidative stress and improve their therapeutic efficacy. Flavonoids precondition and prime MSCs via the direct Keap1-Nrf2 pathway or indirect PI3K-Akt pathway, which enhances cellular resilience to adverse conditions by reducing apoptosis and promoting survival. Primed MSCs, in turn, remodel the microenvironment through an altered secretory profile, releasing bioactive factors that create more favorable conditions for their own persistence.
The core logic of these strategies lies in simulating or inducing adaptive stress, such as employing specific chemical molecules or drug stimuli, or utilizing physical / microenvironmental preconditioning to mimic specific physical conditions of the in vivo injury environment. The most straightforward strategy is overexpression of Nrf2 or its key downstream effector molecules.
The majority of existing studies remain at the level of observing correlations with Nrf2 upregulation, and there is still a lack of precise causal validation regarding key upstream signals – such as specific cytokines, miRNAs, or proteins – through which MSCs or derivatives initiate Nrf2 activation. Mechanistic insights are predominantly derived from in vivo or rodent (mouse/rat) model experiments, with a notable absence of clinical validation, insufficient long-term safety and pharmacokinetic data, and a lack of standardization in administration routes and dosages, all of which hinder clinical translation.
The essential role of the Nrf2 pathway has not been rigorously confirmed, as most studies have not employed reverse genetic validation using Nrf2-knockout animals or specific inhibitors. Consequently, it remains unclear whether therapeutic effects are necessarily and exclusively dependent on Nrf2, and potential synergistic contributions from other pathways may have been overlooked.
Most natural flavonoids face challenges such as low oral bioavailability, rapid metabolism, and poor targeting. Numerous challenges remain to be addressed in order to translate these promising preclinical findings into clinical practice. Future research should focus on the following aspects:
- Elucidating precise upstream molecular mechanisms by which MSCs activate Nrf2;
- Employing more clinically relevant chronic disorder models;
- Systematically evaluating long-term safety, optimal delivery strategies (including dosage and route of administration), and immunogenicity of MSCs-based therapies;
- Validating selection criteria (optimal source), quality control, batch-to-batch consistency of MSCs, and addressing regulatory and ethical barriers to clinical translation; and
- Integrating molecular docking, ADMET (Absorption, Distribution, Metabolism, Excretion, Toxicity) prediction, and in vitro and in vivo validation to further elucidate regulatory effects of flavonoids and enhance understanding of their mechanisms of action.”
https://link.springer.com/article/10.1186/s13287-026-04925-6 “Activation of Nrf2 with natural flavonoids and mesenchymal stromal/stem cells: mechanisms and therapeutic potential for inflammatory diseases” (click pdf)
This paper was overly long at 127 pages, so I focused on the later sections. None of these treatments are currently ready for clinical trials.
I also didn’t mention specific flavonoids as Nrf2 activators. It’s beyond a reviewer’s task to rank Nrf2 activators, and a study’s researchers seldom address why they used a poorly-activating flavonoid instead of a higher-ranked natural plant compound such as sulforaphane.