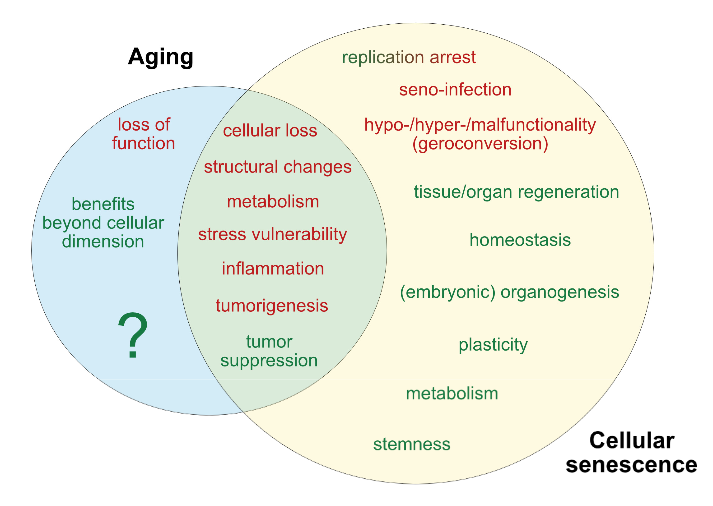
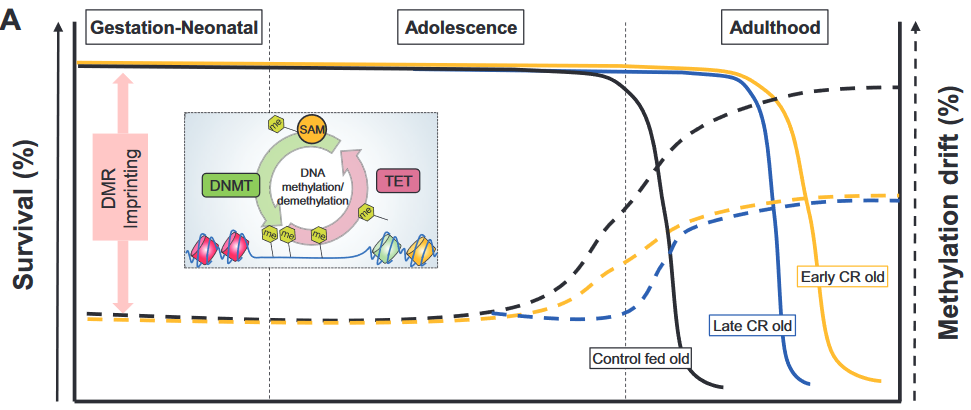
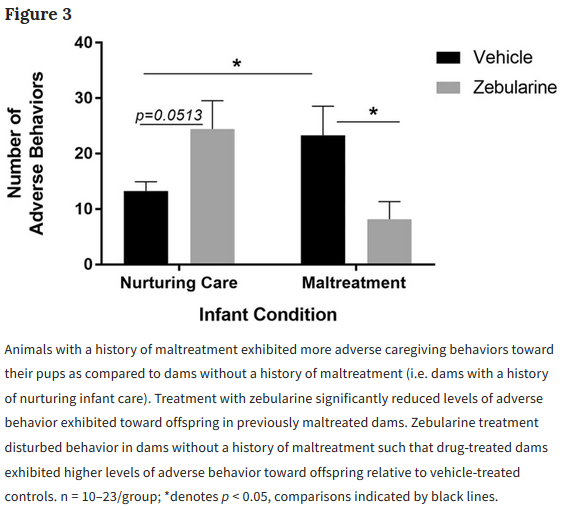
This 2019 McGill rodent study found:
“Prenatal stress exacerbates pain after injury. Analysis of mRNA expression of genes related to epigenetic regulation and stress responses in the frontal cortex and hippocampus, brain structures implicated in chronic pain, showed distinct sex and region-specific patterns of dysregulation.
In general, mRNA expression was most frequently altered in the male hippocampus and effects of prenatal stress were more prevalent than effects of nerve injury. Recent studies investigating chronic pain-related pathology in the hippocampus in humans and in rodent models demonstrate functional abnormalities in the hippocampus, changes in associated behavior, and decreases in adult hippocampal neurogenesis.
The change in expression of epigenetic- and stress-related genes is not a consequence of nerve injury but rather precedes nerve injury, consistent with the hypothesis that it might play a causal role in modulating the phenotypic response to nerve injury. These findings demonstrate the impact of prenatal stress on behavioral sensitivity to a painful injury.

Decreased frontal mRNA expression of BDNF and BDNF IV in male offspring following neuropathic pain or prenatal stress respectively. Relative mRNA expression of other stress-related genes (GR17, FKBP5) and epigenetic-related genes (DNMTs, TETs, HDACs, MBDs, MeCP2) in male offspring.
A drastic decrease in expression of HDAC1 was observed in all groups compared to sham-control animals. CCI: chronic constriction injury.”
The study’s design was similar to the PRS (prenatal restraint stress) model, except that the PRS procedure covered gestational days 11 to 21 (birth):
“Prenatal stress was induced on Embryonic days 13 to 17 by restraining the pregnant dams in transparent cylinder with 5 mm water, under bright light exposure, 3 times per day for 45 min.”
None of the French, Italian, and Swiss PRS studies were cited.
The limitation section included:
- “Although our study shows significant changes in expression of epigenetic enzymes, it didn’t examine the impact of these changes on genes that are epigenetically regulated by this machinery or their involvement in intensifying pain responses.
- The current study is limited by the focus on changes in gene expression which do not necessarily correlate with changes in protein expression.
- Another limitation of this study is the inability to distinguish the direct effects of stress in utero vs. changes in the dam’s maternal behavior due to stress during pregnancy; cross-fostering studies are needed to address this issue.
- Functional experiments that involve up and down regulation of epigenetic enzymes in specific brain regions are required to establish a causal role for these processes in chronic pain.”
What do you think about possible human applicability of this study’s “effects of prenatal stress were more prevalent than effects of nerve injury” finding?
Are there any professional therapeutic frameworks that instruct trainees to recognize that if a person’s mother was stressed while pregnant, their prenatal experiences could cause more prevalent biological and behavioral effects than a recent injury?
https://www.sciencedirect.com/science/article/pii/S0166432819315219 “Prenatal maternal stress is associated with increased sensitivity to neuropathic pain and sex-specific changes in supraspinal mRNA expression of epigenetic- and stress-related genes in adulthood” (not freely available)