This 2019 review of epigenetic clocks by Washington cancer researchers ignored the elephant in the room: Their epigenetic drift paradigm is generally inapplicable to humans because the vast majority of our cells don’t divide/proliferate. They repeatedly returned to an argument for randomness as a cause for aging and disease:
“A time-dependent stochastic event process, like epigenetic drift, could lead to cancer formation through the accumulation of random epigenetic alterations that, through chance, eventually alter epigenetic driver gene expression leading to a clone of cells destined to become cancer.
It is plausible that the stochastic process inherent in epigenetic drift can induce aberrant methylation events that accumulate in normal cells and eventually induce cancer formation.
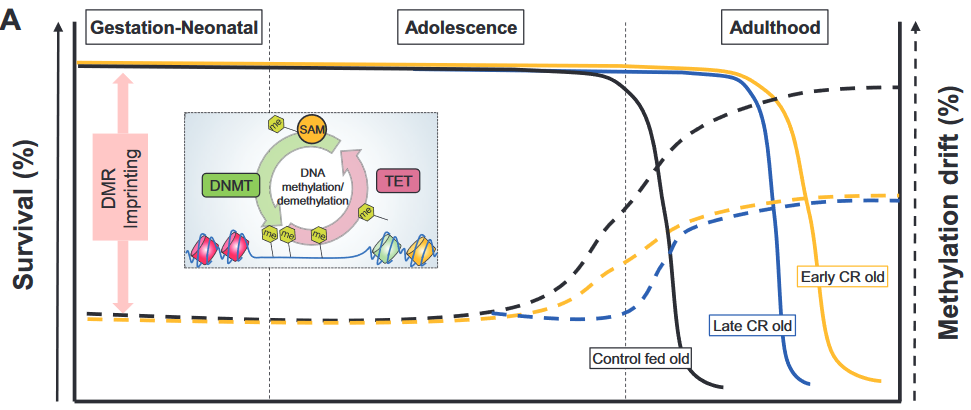
Epigenetic drift relates to a biological process that changes the DNA methylome with age via stochastic gains or losses of DNA methylation. Epigenetic drift can be understood in terms of errors in DNA methylation maintenance during DNA-replication.
The phenomenon of (epi)genetic drift is generally associated with phenotypic neutrality.

For patients who develop cancer around age 80, the most likely initiation time for the founder adenoma cell is predicted to be very early in life, roughly between the ages 15 to 20 years. This unexpected and provocative finding suggests that the optimal age-range for prevention of colorectal cancer may be in adolescence and early adulthood (and ideally through lifelong) dietary and lifestyle interventions.”
The reviewers’ strawman arguments intentionally mischaracterized aspects of the epigenetic clock:
1. The epigenetic clock founder’s actual view on aging was in The epigenetic clock theory of aging:
“The proposed epigenetic clock theory of ageing views biological ageing as an unintended consequence of both developmental programmes and maintenance programmes, the molecular footprints of which give rise to DNAm age estimators.”
The reviewers omitted this intrinsic view of aging, which didn’t fit into the above graphic.
2. Another misrepresentation was:
“In contrast to epigenetic clocks, epigenetic drift refers to a stochastic process that involves both gains and losses of the methylation state of CpG dinucleotides over time.”
A reader of the original 2013 epigenetic clock study would understand that epigenetic clocks measure “both gains and losses of methylation” as in:
“The 193 positively and 160 negatively correlated CpGs get hypermethylated and hypomethylated with age, respectively.”
3. These reviewers omitted recent epigenetic clock significant developments. For example, there was no mention of the GrimAge study, although it was published before this review was submitted.
4. Epigenetic drift as the cause of aging and disease has abundant contrary evidence. These reviewers tossed in a little toward the end of their directed narrative:
“We found only a small number of drift-related CpG island-gene pairs for which drift correlated positively and significantly with gene expression.
The functional consequences of epigenetic drift need to be further elucidated.”
However, they never acknowledged the elephant in the room!
https://cancerres.aacrjournals.org/content/early/2019/11/06/0008-5472.CAN-19-0924 “Epigenetic aging: more than just a clock when it comes to cancer” (not freely available)