I’ll curate this 2019 German review through its figures:
“With the discovery of beneficial aspects of cellular senescence and evidence of senescence being not limited to replicative cellular states, a redefinition of our comprehension of aging and senescence appears scientifically overdue.

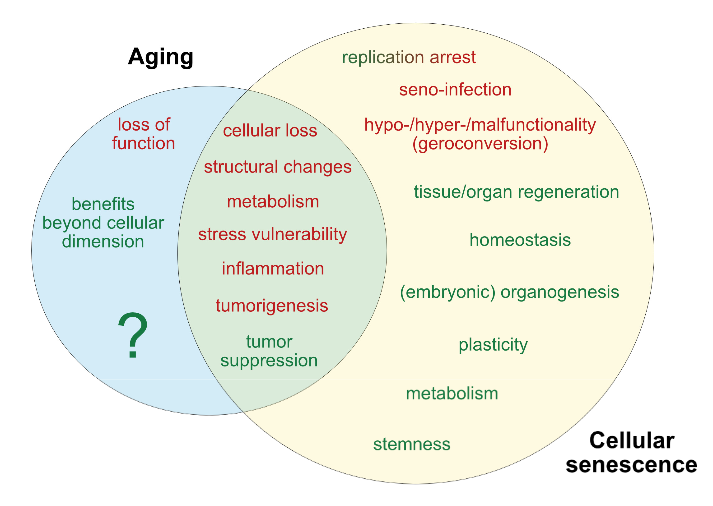
Figure 1. Current determinants and relevant open questions, marking the processes of aging and senescence as discussed in the text. Aspects represented in green are considered as broadly accepted or scientifically consolidated. Novel aspects that are yet unproven, or are under debate, are highlighted in red.
SASP = senescence-associated secretory phenotype. AASP = putative aging-associated secretory phenotype as suggested in the text.

Figure 2. Theories on the causality and purpose of aging. Graphically summarized are four contrasting concepts crystallized from current evidence addressing the inductive driving force of aging. Apart from a stochastic deleteriome, there are arguments for a pseudo-programmed, programmed or at least partially programmed nature of aging.

Figure 3. Comparative representation of the aging and senescence processes highlighting different levels of interaction and putative sites of interventions.
(1) As discussed in the text, causative mechanisms of aging are still not well understood, however, multiple factors including genetic, epigenetic and stress-related effects seem to have an orchestrated role in the progression of aging. Senescence on the other hand, is seen as a programmed response to different kinds of stressors, which proceed in defined stages. Whether, in analogy, aging also follows a defined program or sequential stages is not known.
(2) Senescence involves autocrine and paracrine factors, which are responsible for a ‘seno-infection’ or bystander effect in neighboring cells. There is currently no direct evidence for a similar factor composition propagating the aging process via a kind of ‘gero-infection’.
(3) Accumulation of senescent cells has been described as a hallmark of aging; however, whether they are a causative factor or a consequence of tissue and organismal aging is still unknown. As discussed in the text, it appears possible that aging and senescence mutually influence each other through positive feedback at this level, leading to accelerated tissue damage and aging.
(4,5) Clearance of senescent or aging cells might constitute putative targets for interventional approaches aimed to reduce or reverse the impact of aging and improve cell and tissue homeostasis by inducing a ‘rejuvenation’ process.

Figure 4. Pathological and beneficial functions of aging and senescence, according to current knowledge. In red are represented pathological consequences and in green beneficial functions of aging and senescence.
The impact of aging has mainly been described at the organismal level, since a complete cellular functional profile has not yet been established. Accordingly, whether beneficial consequences of the aging process exist at the cellular level is unclear.”
The assertion of Figure 3 (2) that:
“There is currently no direct evidence for a similar factor composition propagating the aging process via a kind of ‘gero-infection.”
was shown to be false in Reevaluate findings in another paradigm:
“It was demonstrated that increased aging occurred as a result of lack of gonadotropin-releasing hormone and that increased lifespan resulted from its provision during aging.
In this manner:
- Aging of hypothalamic microglia leads to
- Aging of the hypothalamus, which leads to
- Aging elsewhere in the body.
So here we have a multi-level interaction:
- Activation of NF-κB leads to
- Cellular aging, leading to
- A diminished production of GnRH, which then
- Acts (through cells with a receptor for it, or indirectly as a result of changes to GnRH-receptor-possessing cells) to decrease lifespan.
So the age state of hypothalamic cells, at least with respect to NF-κB activation, is communicated to other cells via reduced output of GnRH.”
The reviewers’ position on Figure 2 was:
“In our view, recent evidence that
- Senescence is based on an unterminated developmental growth program and the finding that
- The concept of post-mitotic senescence requires the activation of expansion, or ‘growth’ factors as a second hit,
favor the assumption that aging underlies a grating of genetic determination similarly to what is summarized above under the pseudo-programmed causative approach.”
Their position on Figure 4’s beneficial effects of aging began with the sentence:
“If we assume that aging already starts before birth, it can be considered simply a developmental stage, required to complete the evolutionary program associated with species-intrinsic biological functions such as reproduction, survival, and selection.”
Cited studies included:
https://www.mdpi.com/2073-4409/8/11/1446 “Dissecting Aging and Senescence-Current Concepts and Open Lessons”