This 2020 Swiss/German review mainly cited weed, worm, and yeast studies:
“RNA interference-related mechanisms can mediate the deposition and transgenerational inheritance of specific chromatin modifications in a truly epigenetic fashion.
Epigenetics was initially defined as any heritable change in gene expression patterns without changes in the DNA sequence. Now, epigenetic phenomena are often characterized as ‘gene expression changes that are mutation independent and heritable in the absence of the triggering event’, a definition we will follow in this review. We note that this definition can be expanded to include protein only-based inheritance mechanisms that do not necessarily cause changes in gene expression.
Gene silencing can persist over multiple generations in the germline of C. elegans. Gene repression is typically maintained without the initial trigger for three to seven generations and occasionally for tens of generations. In contrast, silencing of somatically expressed genes mostly affects only the subsequent generation through nonepigenetic parental effects.
In the presence of an ‘enabling’ mutation, primary siRNAs [small interfering RNAs] can trigger an RNAe [RNA-induced epigenetic silencing] response. Secondary siRNA amplification is required for transgenerational inheritance.
The fitness of a population in a dynamic environment strongly depends on the ability of individuals to adapt to the new condition as well as to remember, inherit, and forget such adaptation:
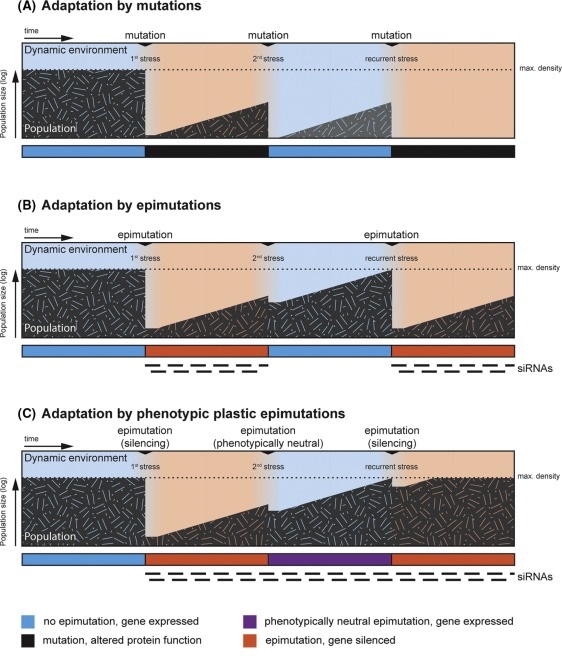
- (A) A well-adapted population (grey) is at its maximal density (dotted line) in a given niche until an environmental change (1st stress) creates a bottleneck. Only few individuals can adapt through mutations and repopulate the niche. After the environment changes back to the initial blue state, only individuals that acquire rare counteracting mutations survive, often leading to extinction of the population.
- (B) Individuals of a population in the red state can gain beneficial epimutations through siRNAs and repopulate the niche. When exposed again to the blue state, the epimutations can be quickly reversed and the population rapidly reaches maximal density. After recurrence of the red state, organisms establish de novo epimutations with the same low frequency as when they first encountered this state.
- (C) In contrast, organisms that can maintain the memory of a beneficial silencing event can quickly re-establish beneficial epimutations and grow to full density. Such memory can be maintained by phenotypically neutral epimutations, marked by the continuously high production of siRNAs without substantial reductions in the expression of a gene. A population that can adapt through phenotypically plastic epimutations is predicted to have a maximal fitness advantage in a dynamic environment.”
The Concluding Remarks section included:
“RNA-mediated epigenetic responses could contribute to adaptation.
Even though RNAe may yield significant adaptive advantages, a high induction frequency could cause silencing of multiple essential genes and therefore be detrimental. Hence, it is plausible that mechanisms would have coevolved that counteract silencing.
Similarly, if constituting a bet-hedging strategy to cope with ever-changing environments, permanent fixation of an acquired silencing response would not constitute a selective advantage and mechanisms that modify and limit the duration of RNAe would be predicted.”
https://www.sciencedirect.com/science/article/pii/S0168952519302598 “Small RNAs in the Transgenerational Inheritance of Epigenetic Information”
The review’s arguments were based on evolutionary selective advantages and less-complex organisms. It predicted that there would be an endpoint generation as in the (A) case of the above graphic.
Were the mechanisms in the (B) case necessarily transgenerational throughout? The review further explained:
“Epimutations tend to occur in hot spots (e.g., in stress-related or nutritional pathway genes) and can potentially silence several homologous genes simultaneously. Incomplete penetrance of a beneficial epimutation by stochastic loss of siRNAs [59] can result in loss of adaptation in a given environment (red state), but can be beneficial if the previous blue state is re-established. However, when the environment changes back to the red state, epimutations must initiate de novo, at the same low frequency as when the population first encountered this state.”
The study cited at 59 found:
“A feedback between siRNAs and RNAi genes determines heritable silencing duration”
but not “Incomplete penetrance of a beneficial epimutation by stochastic loss of siRNAs.” Hmm.
In any event, the review stated:
“Evidence for naturally occurring RNAe-related phenomena in other animals is scarce and we should be cautious about inferring RNAe as a widely conserved phenomenon.”
It’s encouraging to read studies that find benefits to epigenetic transgenerational inheritance, albeit in organisms that are less complex than rodents and humans.