People will forgive you for being wrong, but they will never forgive you for being right – especially if events prove you right while proving them wrong. Thomas Sowell
This 2018 Texas human review subject was prostate cancer epigenetics:
“We comprehensively review the up-to-date roles of epigenetics in the development and progression of prostate cancer. We especially focus on three epigenetic mechanisms: DNA methylation, histone modifications, and noncoding RNAs. We elaborate on current models/theories that explain the necessity of these epigenetic programs in driving the malignant phenotypes of prostate cancer cells.
It is now generally accepted that epigenetics contributes to the development of nearly every stage of PCa [prostate cancer]. Considering the highly heterogeneous nature of PCa, it is quite likely that [the] effect of a particular epigenetic pattern on growth of cancer cells varies from case to case and [is] context specific.
Restoration of a “normal” epigenetic landscape holds promise as a cure for prostate cancer.”
The review’s Epigenetic Therapy section explained much of what’s going on in the above graphic. Its Table 3 was instructive for up-to-date clinical trial information on epigenetic treatments of prostate cancer.
“Restoration of a “normal” epigenetic landscape” won’t guarantee a healthy outcome once diseases start. Prevention seems desirable to avoid the situation where:
“Numerous epigenetic alterations reinforce the establishment of a context-specific transcriptional profile that favors self-renewal, survival, and invasion of PCa cells.”
This 2018 Canadian cell study described the development of a single-cell protocol to:
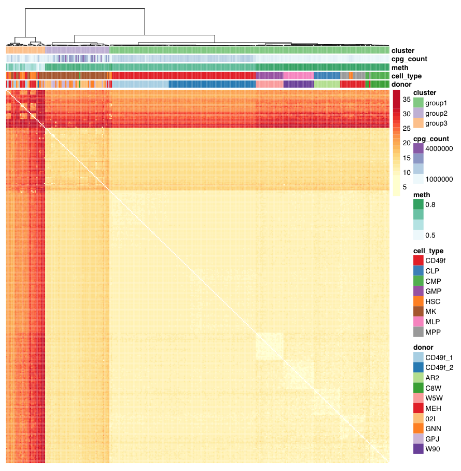
“Profile primitive hematopoietic cells of mouse and human origin to identify epigenetically distinct subpopulations. Deep sampling of the CpG content of individual HSCs allowed for the near complete reconstitution of regulatory states from epigenetically defined subpopulations of HSCs and revealed a high level of redundancy of CpG methylation states within these phenotypically defined hematopoietic cell types.
Hematopoietic stem cells (HSCs) are functionally defined cells that display evidence of extensive self-renewal of their ability to generate mature blood cells for the lifetime of the organism and following transplantation into myelosuppressed permissive hosts. Most of the epigenetic measurements underpinning these observations represent consensus values experimentally derived from thousands of cells partially enriched in HSCs or their progeny, thus failing to discern distinct epigenetic states within HSCs.
Current analytical strategies for single-cell DNA methylation measurements average DNA methylation in fixed genomic bins or over defined genomic regions.
However, inference across cells (as well as sequence context) assumes homogeneity across cells, which is at cross-purposes with the generation of single-cell molecular measurements through the potential to mask rare subpopulations.
We identified donor as a significant source of consistent epigenetic heterogeneity, which was reduced but not eliminated by correcting for personal genetic variants. This observation is consistent with previous reports that showed genetic diversity as related to but not accountable for all DNA methylation differences and suggests that in utero environmental differences may be encoded within the HSC compartment.”
The study advanced science not only by measuring CpG methylation within each HSC, but also by producing another data point “that in utero environmental differences may be encoded within the HSC compartment.”
The paragraph with “assumes homogeneity across cells” bold text provided another example of the statistical analysis flaw that gives individually inapplicable results per Group statistics don’t necessarily describe an individual. The above graphic of human hematopoietic phenotypes demonstrated that the researchers have potentially solved this problem by measuring individual cells.
The researchers discussed another aspect of the study that’s similar to the epigenetic clock methodology:
“Phenotype-specific methylation signatures are characterized by extensive redundancy such that distinct epigenetic states can be accurately described by only a small fraction of single-CpG methylation states. In support of such a notion, the unique components of a DNA methylation “age” signature are contained in ∼353 CpGs sites, presumably representing a random sample of a total age signature that involves many more sites not detected using the reduced representation strategies from which these signatures have been derived.”
Also, in The epigenetic clock theory of aging the originator of the epigenetic clock characterized HSCs as an effective intervention against epigenetic aging:
“In vivo, haematopoietic stem cell therapy resets the epigenetic age of blood of the recipient to that of the donor.”
This 2018 German review provided short summaries of 44 studies on the contribution of histone modifications to allergies. An overall summary of their search results was:
“There are at least two levels at which the role of histone modifications is manifested.
One is the regulation of cells that contribute to the allergic inflammation (T cells and macrophages) and those that participate in airway remodeling.
The other is the direct association between histone modifications and allergic phenotypes.
Inhibitors of histone-modifying enzymes may potentially be used as anti-allergic drugs. Furthermore, epigenetic patterns may provide novel tools in the diagnosis of allergic disorders.”
This type of search is what’s expected of researchers who will perform either:
A meta-analysis of studies selected from the search results; or
Their own study.
These reviewers didn’t indicate that they were proceeding along either path.
The review was fine for the purpose of presenting current studies of the subject. But this was just the preparatory stage of research.
This 2018 German review was comprehensive for its subject, epigenetic control of variation and stochasticity in metabolic disease. I’ll focus on one aspect, phenotypic variation:
“Phenotypic [Mendelian] variation can result both from gain- and loss-of-function mutations. Because of the extreme interconnectivity of cell regulatory networks, even at the cellular level, predicting the impact of a sequence variant is difficult as the resultant variation acts:
In the context of all other variants and
Their potential additive, synergistic and antagonistic interactions.
This phenomenon is known as epistasis.
∼98.5% of our genome is non-protein-coding: it is pervasively transcribed, and its transcripts can support regulatory function. Among the best functionally characterized non-coding RNAs (ncRNAs) arising from these sequences are microRNAs (miRNAs).
Environmental [non-Mendelian] variation or ‘stimuli’ occurring during critical windows of susceptibility can elicit lifelong alterations in an individual’s phenotype. Intergenerational metabolic reprogramming [in fruit flies] results from global alterations in chromatin state integrity, particularly from reduced H3K27me3 and H3K9me3 [histone] domains.
The broad variation of fingerprints in humans is thought to depend to a large degree on stochastic variation in mechanical forces. These clear examples of inducible multi-stable or stochastic variation highlight how little we know about the landscape of potential phenotypic variation itself.
Consensus estimates of heritability for obesity and T2D are ∼70% and ∼35% respectively. The remaining, unexplained component is known to involve gene–environment interactions as well as non-Mendelian players.”
Although the above graphic displays transgenerational inheritance for humans, the reviewers didn’t cite any human studies that adequately demonstrated causes for and effects of transgenerational epigenetic inheritance.
I’ve read the cited Swedish and Dutch studies. Their designs, methods, and “correlate with” / “was associated with” results didn’t provide incontrovertible evidence from the F0 great-grandparents, F1 grandparents, F2 parents, and F3 children. It’s necessary to thoroughly study each generation to confirm definitive transgenerational epigenetic inheritance causes and effects.
As noted in How to hijack science: Ignore its intent and focus on the 0.0001%, there aren’t any such published studies to cite. Researchers urgently need to do this human research, and stop using these poor substitutes [1] to pretend there are already adequately evidenced transgenerational epigenetic inheritance human results.
I downgraded the review for treating research of this and other subjects as faits accomplis. It’s opposite ends of the evidential spectrum to state “how little we know about the landscape of potential phenotypic variation,” and in the same review, speciously extrapolate animal experiments into putative human results.
[1] As an example of the poor substitutes for evidence, a researcher referred me to the 2013 “Transgenerational effects of prenatal exposure to the 1944–45 Dutch famine” which is freely available at https://obgyn.onlinelibrary.wiley.com/doi/full/10.1111/1471-0528.12136 as a study finding human transgenerational epigenetic inheritance.
The Methods section showed:
The study’s non-statistical data was almost all unverified self-reports by a self-selected sample of the F2 generation, average age 37.
No detailed physical measurements or samples were taken of them, nor of the F1 generation, nor of the F0 generation, all of which are required as baselines for any transgenerational epigenetic inheritance findings.
No detailed physical measurements or samples were taken of the F3 generation, which is the generation that may provide transgenerational evidence if the previous generations also have detailed physical baselines.
The study’s researchers drew enough participants (360) such that their statistics package allowed them to impute and assume into existence a LOT of data. But the scientific method constrained them to make factual statements of what the evidence actually showed. They admitted:
“In conclusion, we did not find a transgenerational effect of prenatal famine exposure on the health of grandchildren in this study.”
Yet this study is somehow cited for evidence of human transgenerational epigenetically inherited causes and effects!
This 2018 Michigan human cell study subject was factors affecting the expression of human endogenous retroviruses:
“We provide a comprehensive genomic and epigenomic map of the more than 500,000 endogenous retroviruses (ERVs) and fragments that populate the intergenic regions of the human genome.
The repressive epigenetic marks associated with the ERVs, particularly long terminal repeats (LTRs), show a remarkable switch in silencing mechanisms, depending on the evolutionary age of the LTRs:
Young LTRs tend to be CpG-rich and are mainly suppressed by DNA methylation, whereas
Intermediate age LTRs are associated predominantly with histone modifications, particularly histone H3 lysine 9 (H3K9) methylation.
The evolutionarily old LTRs are more likely inactivated by the accumulation of loss-of-function genetic mutations.
Because the expression of ERVs is potentially dangerous to the host cell, understanding the repressive mechanisms is important. Earlier studies have implicated the aberrant expression of ERVs in autoimmune disease pathogenesis. However, this “enemy within” may also play a beneficial role in cancer therapy.
The same kinds of chromatin dynamics appear to be used both by LTRs and genes.”
I wasn’t going to curate this study before I saw the above graphic of our Boreoeutherian ancestor. Evolutionary subjects seem very abstract until an artist reconstructs the data visually.
This post has somehow become a target for spammers, and I’ve disabled comments. Readers can comment on other posts and indicate that they want their comment to apply here, and I’ll re-enable comments.
This 2018 Oregon rodent study fed a 15% broccoli sprout diet beginning at four weeks of age to a mouse strain with a near-100% chance of developing prostate cancer:
“Broccoli sprouts reduced prostate cancer incidence and progression to invasive cancer. Broccoli sprout consumption also decreased histone H3 lysine 9 trimethylation in the ventral lobe (age 12 wk), and decreased histone H3 lysine 18 acetylation in all prostate lobes (age 28 wk).
The TRAMP model of prostate cancer was utilized because the tumors occur in the prostate epithelium and the tumor tissue histopathology closely mimics human disease. Additional advantages include that the tumors arise spontaneously and appear in ∼100% of mice.”
“This 15% broccoli sprout diet had 400 mg SFN [sulforaphane]/kg diet, which was chosen because it is equivalent to 1 mg SFN/d which has been used in previous studies.
Food consumption was measured over the course of the study and no difference was found in the intake of food between the control and broccoli sprout–fed groups.”
To be “equivalent to 1 mg SFN/d” at a .4 mg sulforaphane/gram rate, the animals would eat 2.5 grams per day. That’s half of a normal intake. “Food consumption was measured” but not disclosed.
“4 week old male TRAMP mice were treated with PBS [phosphate-buffered saline] (control) or 1 mg SFN in PBS three times/week for 15-18 weeks.”
not “1 mg SFN/d which has been used in previous studies.”
The researchers didn’t sufficiently quantify their findings to help humans, which is the basic purpose of any animal study.
https://academic.oup.com/cdn/article/2/3/nzy002/4803105 “Broccoli Sprouts Delay Prostate Cancer Formation and Decrease Prostate Cancer Severity with a Concurrent Decrease in HDAC3 Protein Expression in Transgenic Adenocarcinoma of the Mouse Prostate (TRAMP) Mice”
This 2018 Italian human cell study conducted a series of experiments on the effects of nutrient deprivation:
“Reduced food intake, and in particular protein or amino acid (AA) restriction, extends lifespan and healthspan.
We have previously shown that, in mammalian cells, deprivation of essential AAs (methionine/cysteine or tyrosine) leads to the transcriptional reactivation of integrated silenced transgenes by a process involving epigenetic chromatic remodeling and histone acetylation.
Here we show that the deprivation of methionine/cysteine also leads to the transcriptional upregulation of endogenous retroviruses [ERVs], suggesting that essential AA starvation affects the expression not only of exogenous non-native DNA sequences, but also of endogenous anciently-integrated and silenced parasitic elements of the genome.
ERVs, comprising 8% of the human genome, represent the remnants of past infections of germ cells by exogenous retroviruses, and are mostly unable to retrotranspose in the human genome. However, they can reactivate during physiological development, or in pathological conditions like cancer, and regulate the expression of nearby genes by their LTR elements, leading to general transcriptional reprogramming.
Dissection of the underlying mechanism ruled out a role for the main AA-deficiency sensor GCN2 and pointed to the ribosome as the possible master controller.”
Contrary to what’s implied by its title, though, and as I noted in How to hijack science: Ignore its intent and focus on the 0.0001%, those reviewers didn’t cite any human studies that adequately demonstrated transgenerational epigenetic inheritance causes and effects. They admitted:
“Direct evidence that epigenetic factors drive the inheritance of T2DM [type 2 diabetes mellitus] in humans is lacking.”
The Danish reviewers then continued on as if proof of human transgenerational epigenetic inheritance was a foregone conclusion! It didn’t serve any valid scientific purpose to assume such evidence into existence.
This 2018 Belgian review subject was in part the transgenerational epigenetic effects of maternal obesity during pregnancy. The subject was tailored for the journal in which it appeared, Atherosclerosis, so other transgenerationally inherited epigenetic effects weren’t reviewed:
“The transgenerational impact of these alterations in methylation patterns are only shown in animal studies with HFD [high-fat diet] animals. In this respect the paternal influence also comes forward.
Alterations in methylation at the spermatozoa of male rats fed with a HFD were shown in combination with transgenerational metabolic effects, mainly on the female offspring. Methylation alterations in spermatozoa were also found in the male offspring of dams fed with HFD during their pregnancy. Consequent effects on the phenotype were again only shown in female offspring (until third generation).
A transgenerational inheritance through the female germline by mitochondrial inheritance has been suggested. A recent, small study in humans found altered mitochondrial functioning in the male offspring of overweight woman. A finding that has been confirmed in mice studies with a persistence of this transfer of aberrant oocyte mitochondria into the third generation.
The identification of a number of alterations in active cardiovascular microRNA species in the offspring of animals with obesity offer promising perspectives for the future.”
Evidence for transgenerational aspects of in uteroprogramming included two studies I hadn’t previously curated:
This 2018 Polish review subject was relationships between melatonin and depression:
“Although melatonin has been known about and referred to for almost 50 years, the relationship between melatonin and depression is still not clear. In this review, we summarize current knowledge about genetic and epigenetic regulation of enzymes involved in melatonin synthesis and metabolism as potential features of depression pathophysiology and treatment.
Melatonin has an antidepressant effect by:
Maintaining the body’s circadian rhythm;
Regulating the pattern of expression of clock genes in the suprachiasmatic nucleus (SCN); and
Modifying key genes of serotoninergic neurotransmission that are linked with a depressive mood.
Light input causes release of γ-aminobutyric acid (GABA) by the SCN, and the inhibitory signal is transmitted to the pineal gland to inhibit melatonin production.
Melatonin is produced via metabolism of serotonin in two steps which are catalyzed by serotonin N-acetyltransferase (SNAT) and acetylserotonin-O-methyltransferase (ASMT). Serotonin, SNAT, and ASMT are key melatonin level regulation factors.
Both melatonin and serotonin are synthesized from the same amino acid, tryptophan. People on a high tryptophan diet (>10 mg/kg body weight per day) have a significantly lower level of depressive symptoms, irritation, and anxiety than people on a low tryptophan diet (<5 mg/kg body weight per day).
To our knowledge, there are only 2 studies in the literature that characterize mRNA expression of ASMT in the peripheral blood of recurrent depressive disorders. They demonstrated reduced mRNA expression of ASMT in patients with depression and cognitive impairment. Surprisingly, these studies, despite promising results, have not been replicated. Moreover, no analysis of other melatonin related-genes as potential biomarkers of depression has been provided.
The main monoamine hypothesis of pathophysiology of depression indicates that depression is induced by a change in levels of ≥1 monoamines such as serotonin, noradrenaline, and dopamine. Evidence for the serotonergic theory is an observation that antidepressants such as tricyclic antidepressants, selective serotonin reuptake inhibitors, and noradrenaline reuptake inhibitors increase the level of serotonin in the brain.
We focus on serotonin as a neurotransmitter which is a precursor of melatonin synthesis. In a depressed patient, serotonin synthesis is impaired, and poor precursor availability may prevent formation of an adequate amount of melatonin. However, only a few studies have analyzed the relationship between serotonin and melatonin levels and the correlation with blood serum.”
At eight cents a day ($.04 for women), melatonin is a cheap and effective supplement.
I hadn’t considered possible antidepressant effects until reading this review. More human studies are needed.
The principal way science advances is through a principle Einstein expressed as:
“No amount of experimentation can ever prove me right; a single experiment can prove me wrong.”
The scientific community and public should be satisfied that the scientific process is working well when hypotheses are discarded due to nonconfirming evidence. Researchers should strive to develop evidence that rejects paradigms, and be lauded for their efforts.
The opposite took place with this 2018 commentary on two studies where evidence didn’t confirm current biases. I curated one of these studies in DNA methylation and childhood adversity.
Commentators’ dismissive tone was set in the opening paragraph:
“Is early exposure to adversity associated with a genetic or an epigenetic signature? At first glance, two articles in this issue -..and the other from Marzi et al., who measured genome-wide DNA methylation in a prospective twin cohort assessed at age 18 – appear to say that it is not.”
Commentators – one of whom was a coauthor of Manufacturing PTSD evidence with machine learning, – went on to protect their territory. Nevermind these two studies’ advancement of science that didn’t coincide with commentators’ vested interests.
My main concern with the curated study was that although child subjects had been studied at ages 5, 7, 10, 12, and 18, parents had never been similarly evaluated! Those researchers passed up an opportunity to develop parents as a F0 generation for understanding possible human transgenerational inherited epigenetic causes and effects.
That study focused on the children’s intergenerational epigenetic effects. However, animal studies have often demonstrated transgenerational effects that skip over F1 generation children! For example:
A study not cited in – but completely appropriate for – The lack of oxygen’s epigenetic effects on a fetus found heart disease effects in the F1 generation that were different from the heart disease effects found in F2 and F3 generations.
This 2018 Korean review discussed aspects of the hypothalamus and aging:
“A majority of physiological functions that decline with aging are broadly governed by the hypothalamus, a brain region controlling development, metabolism, reproduction, circadian rhythm, and homeostasis. In addition, the hypothalamus is poised to connect the brain and the body so that the environmental information affecting aging can be transmitted through the hypothalamus to affect the systematic aging of the peripheral organs.
The hypothalamus is hypothesized to be a primary regulator of the process of aging of the entire body. This review aims to assess the contribution of hypothalamic aging to the age-related decline in body functions, particularly from the perspective of:
energy homeostasis,
hormonal balance,
circadian rhythm, and
reproduction,
and to highlight its underlying cellular mechanisms with a focus on:
The reviewers didn’t consider aging to be an “unintended consequence” of development. This perspective was found in a reference to A study of DNA methylation and age:
“Aging is not programmed. Instead, aging is a continuation of developmental growth, driven by genetic pathways.
Genetic programs determine developmental growth and the onset of reproduction. When these programs are completed, they are not switched off.
Aging has no purpose (neither for individuals nor for group), no intention. Nature does not select for quasi-programs. It selects for robust developmental growth.”
“The proposed epigenetic clock theory of ageing views biological ageing as an unintended consequence of both developmental programmes and maintenance programmes.”
“The hypothalamus is hypothesized to be a primary regulator of the process of aging.”
Almost all of the details discussed were from rodent studies.
As detailed in How to cure the ultimate causes of migraines? and its references, the hypothalamus is a brain structure that lacks feedback mechanisms for several of its activities. This structure develops shortly after conception and has an active prenatal role.
The hypothalamus plays its part in getting us developed and ready to reproduce, with certain feedback loops being evolutionarily unnecessary. The hypothalamus perfectly illustrates the point of:
“When these programs are completed, they are not switched off.”
Evolutionarily unnecessary feedback for aspects of hypothalamic activity may result in it not winding down when its developmental role is over. This activity shouldn’t be interpreted to construe a role that has some other meaning or purpose.
This 2018 Loma Linda review subject was gestational hypoxia:
“Of all the stresses to which the fetus and newborn infant are subjected, perhaps the most important and clinically relevant is that of hypoxia. This review explores the impact of gestational hypoxia on maternal health and fetal development, and epigenetic mechanisms of developmental plasticity with emphasis on the uteroplacental circulation, heart development, cerebral circulation, pulmonary development, and the hypothalamic-pituitary-adrenal axis and adipose tissue.
An understanding of the specific hypoxia-induced environmental and epigenetic adaptations linked to specific organ systems will enhance the development of target-specific inhibition of DNA methylation, histone modifications, and noncoding RNAs that underlie hypoxia-induced phenotypicprogramming of disease vulnerability later in life.
A potential stumbling block to these efforts, however, relates to timing of the intervention. The greatest potential effect would be accomplished at the critical period in development for which the genomic plasticity is at its peak, thus ameliorating the influence of hypoxia or other stressors.
With future developments, it may even become possible to intervene before conception, before the genetic determinants of the risk of developing programmed disease are established.”
Table 3 “Antenatal hypoxia and developmental plasticity” column titles were Species | Offspring Phenotypes of Disorders and Diseases | Reference Nos.
This review was really an ebook, with 94 pages and 1,172 citations in the pdf file. As I did with Faith-tainted epigenetics, I read it with caution toward recognizing 1) the influence of the sponsor’s biases, 2) any directed narrative that ignored evidence contradicting the narrative, and 3) any storytelling.
One review topic that was misconstrued was transgenerational epigenetic inheritance of hypoxic effects. The “transgenerational” term was used inappropriately by several of the citations, and no cited study provided evidence for gestational hypoxic effects through the F3 great-grandchild generation.
“One substance that fetuses are frequently exposed to is caffeine, which is a non-selective adenosine receptor antagonist. We discovered that in utero alteration in adenosine action leads to adverse effects on embryonic and adult murine hearts. We find that cardiac A1ARs [a type of adenosine receptor] protect the embryo from in utero hypoxic stress, a condition that causes an increase in adenosine levels.
After birth in mice, we observed that in utero caffeine exposure leads to abnormal cardiac function and morphology in adults, including an impaired response to β-adrenergic stimulation. Recently, we observed that in utero caffeine exposure induces transgenerational effects on cardiac morphology, function, and gene expression.”
The timing of in utero caffeine treatment leads to differences in adult cardiac function, gene expression, and phenotype. Exposure to caffeine from E6.5–9.5 leads the F1 generation to develop dilated cardiomyopathy with decrease % FS and increased Myh7 expression. In utero caffeine exposure from E10.5–13.5 leads to a hypertrophic cardiomyopathy in the F2 generation along with increased % FS and decreased Myh7 expression
Why was this review and its studies omitted? It was on target for both gestational hypoxia and transgenerational epigenetic inheritance of hypoxic effects!
It was alright to review smoking, cocaine, methamphetamine, etc., but the most prevalent drug addiction – caffeine – couldn’t be a review topic?
The Loma Linda review covered a lot, but I had a quick trigger due to the sponsor’s bias. I started to lose “faith” in the reviewers after reading the citation for the review’s last sentence that didn’t support the statement.
My “faith” disappeared after not understanding why a few topics were misconstrued and omitted. Why do researchers and sponsors ignore, misrepresent, and not continue experiments through the F3 generation to produce evidence for and against transgenerational epigenetic inheritance? Where was the will to follow evidence trails regardless of socially acceptable beverage norms?
The review acquired the taint of storytelling with the reviewers’ assertion:
“..timing of the intervention. The greatest potential effect would be accomplished at the critical period in development for which the genomic plasticity is at its peak, thus ameliorating the influence of hypoxia or other stressors.”
Contradictory evidence was in the omitted caffeine study’s graphic above which described two gestational critical periods where an “intervention” had opposite effects, all of which were harmful to the current fetus’ development and/or to following generations. Widening the PubMed link’s search parameters to “caffeine hypoxia” and “caffeine pregnancy” returned links to human early life studies that used caffeine in interventions, ignoring possible adverse effects on future generations.
This is my final curation of any paper sponsored by this institution.
This 2018 Michigan review subject was cancer evolution:
“Based on the fact that cancer typically represents a complex adaptive system, where there is no linear relationship between lower-level agents (such as each individual gene mutation) and emergent properties (such as cancer phenotypes), we call for a new strategy based on the evolutionary mechanism of aneuploidy [abnormal number of chromosomes] in cancer, rather than continuous analysis of various individual molecular mechanisms.
Cancer evolution can be understood by the dynamic interaction among four key components:
Internal and external stress;
Elevated genetic and non-genetic variations (either necessary for cellular adaptation or resulting from cellular damages under stress);
Genome-based macro-cellular evolution (genome replacement, emergent as new systems); and
Multiple levels of system constraint which prevent/slow down cancer evolution (from tissue/organ organization to the immune system interaction).
Since the sources of stress are unlimited and unavoidable (as they are required by all living systems), there are large numbers of gene mutations / epigenetic events / chromosomal aberrations, such as aneuploidy, that can be linked to stress-mediated genomic variants. Furthermore, as environmental constraints are constantly changing, even identical instances of aneuploidy will have completely different outcomes in the context of cancer evolution, as the results of each independent run of evolution will most likely differ.
Most current research efforts are focusing on molecular profiles based on an average population, and outliers are eliminated or ignored, either by the methods used or statistical tools. The traditional view of biological research is to identify patterns from “noise,” without the realization that the so-called “noise” in fact is heterogeneity, which represents a key feature of cancer evolution by functioning as the evolutionary potential.
Understanding the molecular mechanism (both cause and effect) of aneuploidy is far from enough. A better strategy is to monitor the evolutionary process by measuring evolutionary potential. For example, the overall degree of CIN [chromosome instability] is more predictive than individual gene mutation profile.”
Although I read many abstracts of cancer research papers every week, I usually don’t curate them. I curated this paper because the reviewers emphasized several themes of this blog, including:
Further examples of how stress may shape one’s life.
How researchers miss information when they ignore or process away variation:
“Studies have demonstrated the importance of outliers in cancer evolution, as cancer is an evolutionary game of outliers. While this phenomenon can provide a potential advantage for cellular adaptation, it can also, paradoxically, generate non-specific system stress, which can further produce more genetic and non-genetic variants which favor the disease condition.”
Epigenetics researchers may benefit from evolutionary viewpoints that incorporate the interactions of stress and “genetic and non-genetic variants.”
Since epigenetic changes require inheritance in order to persist, it would be a step forward to see researchers start “measuring evolutionary potential” of these inheritance processes.
This 2018 French review subject was mechanisms of autoimmunity:
“Autoimmune diseases (AIDs) encompass more than 80 distinct chronic disorders characterized by inflammatory reactions that can either be systemic or organ specific. In all cases, the disease development is the consequence of the effects of environmental factors in predisposed individuals.
Most of the genes identified by genome-wide association studies (GWAS) on AIDs are related to immunity. However, functional immune parameters that are commonly dysregulated in AIDs do not necessarily stem from these genetic variants. Rather than performing even larger GWAS, understanding complex traits, such as human diseases, may require meticulous analysis or cell-specific gene networks and take into account not only core genes but also seemingly irrelevant genes that may overall have an impact on the disease.
Treg cell defects have been considered a primary cause of AIDs. However, one could ask whether the Treg cell dysfunction exists before the onset of the disease or is provoked by the inflammatory event induced by the triggering components. The defect of Treg cells generally coexists with the inflammatory processes, suggesting several hypotheses:
The inflammation might develop because of a poor regulation of the immune system,
The Treg cells could become inefficient because of the inflammatory environment, or
A common factor concomitantly leads to both effects.
It is likely that autoimmunity results from a chronic imbalance involving both environmental and intrinsic factors. It is now clear that polygenic explanations did not fulfill expectations and that more efforts are needed to understand how the interplay of environmental clues may have a phenotypic impact.”