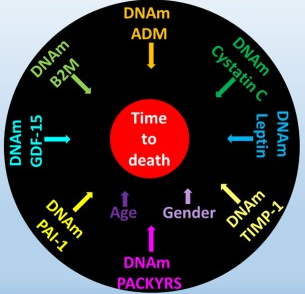
A 2019 UCLA study introduced a derivative of the epigenetic clock named GrimAge:
“DNAm GrimAge, a linear combination of chronological age, sex, and DNAm-based surrogate biomarkers for seven plasma proteins and smoking pack-years, outperforms all other DNAm-based biomarkers, on a variety of health-related metrics.
An age-adjusted version of DNAm GrimAge, which can be regarded as a new measure of epigenetic age acceleration (AgeAccelGrim), is associated with a host of age-related conditions, lifestyle factors, and clinical biomarkers. Using large scale validation data from three ethnic groups, we demonstrate that AgeAccelGrim stands out among pre-existing epigenetic clocks in terms of its predictive ability for time-to-death, time-to-coronary heart disease, time-to-cancer, its association with computed tomography data for fatty liver/excess fat, and early age at menopause.”
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6366976/ “DNA methylation GrimAge strongly predicts lifespan and healthspan”
A miserable attempt at reporting the study’s findings included angles of superstition, fear-of-the-future, and suspicion-by-spurious-association:
“The research has already captured the attention of the life insurance industry. After all, a solid death date could mean real savings when it comes to pricing policies.
The hope is that if and when legitimate anti-aging drugs are developed, GrimAge could be used to test their effectiveness. In a world with functional anti-aging drugs, “doctors could test [your GrimAge number] and say, ‘You know what, you’re aging too quickly. Take this,'” Horvath said.”
https://onezero.medium.com/a-new-test-predicts-when-youll-die-give-or-take-a-few-years-2d08147c8ea6 “A New Test Predicts When You’ll Die (Give or Take a Few Years)”
A detailed blog post from Josh Mitteldorf provided scientific coverage of the study:
“Methylation sites associated with smoking history predicted how long the person would live more accurately than the smoking history itself. Even stranger, the methylation marks most closely associated with smoking were found to be a powerful indication of future health even when the sample was confined to non-smokers.
The DNAm GrimAge clock was developed in two stages, a correlation of a correlation. Curiously, the indirect computation yields the better result.
Horvath’s finding that secondary methylation indicators are more accurate than the underlying primary indicator from which they were derived is provocative, and calls out for a new understanding.”
https://joshmitteldorf.scienceblog.com/2019/03/05/dnam-grimage-the-newest-methylation-clock “DNAm GrimAge—the Newest Methylation Clock”
When there are logical disconnects in findings like the above, it’s time to examine underlying premises. As noted in Group statistics don’t necessarily describe an individual, an assumption required by statistical analyses is that each measured item in the sample is interchangeable with the next.
This presumption is often false, producing individually inapplicable results. For example, Immune memory vs. immune adaptation included this description of the adaptive immune system:
“To be effective, highly specific immune response requires huge diversity of receptors and antibodies, which is achieved by somatic rearrangement of gene segments. Recombination results in millions of TCR [T cell receptor] and antibody variants able to recognize and neutralize millions of various antigens.”
Standard statistics of millions of T cell receptor and antibody variants won’t represent their individually unique properties. But individual differences are both their purpose and benefit to us.
The GrimAge study’s overreach was most apparent in stratifying educational attainment to develop correlations. As mentioned in Does a societal mandate cause DNA methylation? such statistics are poor evidence of each individual’s biological realities.
Neither derivatives of group statistics, nor correlations of correlations, seem to be the techniques needed to understand biological causes of effects. Another commentary on the GrimAge study mentioned but glossed over this point:
“It remains a mystery why exactly the epigenetic clocks work, and whether age-related changes in DNA methylation contribute to the cause of aging or are a result of it.”