This 2018 Canadian cell study described the development of a single-cell protocol to:
“Profile primitive hematopoietic cells of mouse and human origin to identify epigenetically distinct subpopulations. Deep sampling of the CpG content of individual HSCs allowed for the near complete reconstitution of regulatory states from epigenetically defined subpopulations of HSCs and revealed a high level of redundancy of CpG methylation states within these phenotypically defined hematopoietic cell types.
Hematopoietic stem cells (HSCs) are functionally defined cells that display evidence of extensive self-renewal of their ability to generate mature blood cells for the lifetime of the organism and following transplantation into myelosuppressed permissive hosts. Most of the epigenetic measurements underpinning these observations represent consensus values experimentally derived from thousands of cells partially enriched in HSCs or their progeny, thus failing to discern distinct epigenetic states within HSCs.
Current analytical strategies for single-cell DNA methylation measurements average DNA methylation in fixed genomic bins or over defined genomic regions.
However, inference across cells (as well as sequence context) assumes homogeneity across cells, which is at cross-purposes with the generation of single-cell molecular measurements through the potential to mask rare subpopulations.
We identified donor as a significant source of consistent epigenetic heterogeneity, which was reduced but not eliminated by correcting for personal genetic variants. This observation is consistent with previous reports that showed genetic diversity as related to but not accountable for all DNA methylation differences and suggests that in utero environmental differences may be encoded within the HSC compartment.”
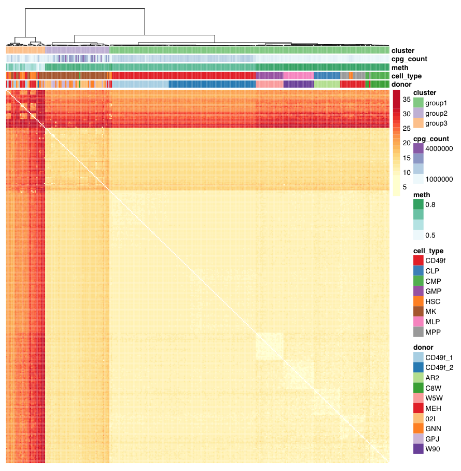
The study advanced science not only by measuring CpG methylation within each HSC, but also by producing another data point “that in utero environmental differences may be encoded within the HSC compartment.”
The paragraph with “assumes homogeneity across cells” bold text provided another example of the statistical analysis flaw that gives individually inapplicable results per Group statistics don’t necessarily describe an individual. The above graphic of human hematopoietic phenotypes demonstrated that the researchers have potentially solved this problem by measuring individual cells.
The researchers discussed another aspect of the study that’s similar to the epigenetic clock methodology:
“Phenotype-specific methylation signatures are characterized by extensive redundancy such that distinct epigenetic states can be accurately described by only a small fraction of single-CpG methylation states. In support of such a notion, the unique components of a DNA methylation “age” signature are contained in ∼353 CpGs sites, presumably representing a random sample of a total age signature that involves many more sites not detected using the reduced representation strategies from which these signatures have been derived.”
Also, in The epigenetic clock theory of aging the originator of the epigenetic clock characterized HSCs as an effective intervention against epigenetic aging:
“In vivo, haematopoietic stem cell therapy resets the epigenetic age of blood of the recipient to that of the donor.”
https://www.cell.com/stem-cell-reports/article/S2213-6711(18)30308-4/fulltext “High-Resolution Single-Cell DNA Methylation Measurements Reveal Epigenetically Distinct Hematopoietic Stem Cell Subpopulations”
