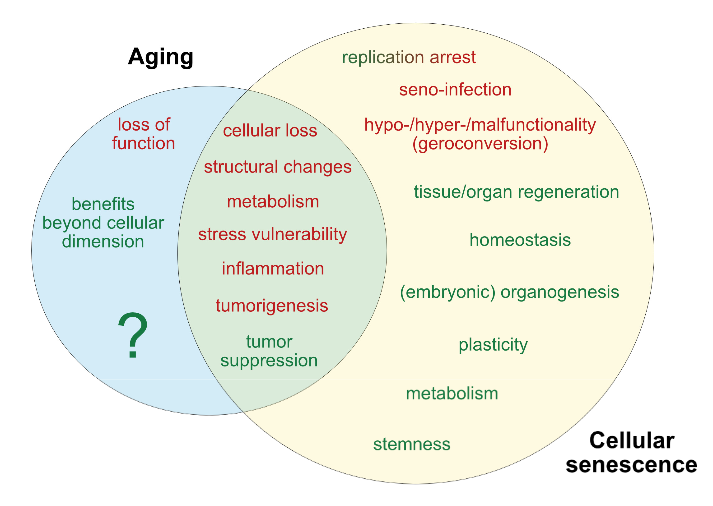
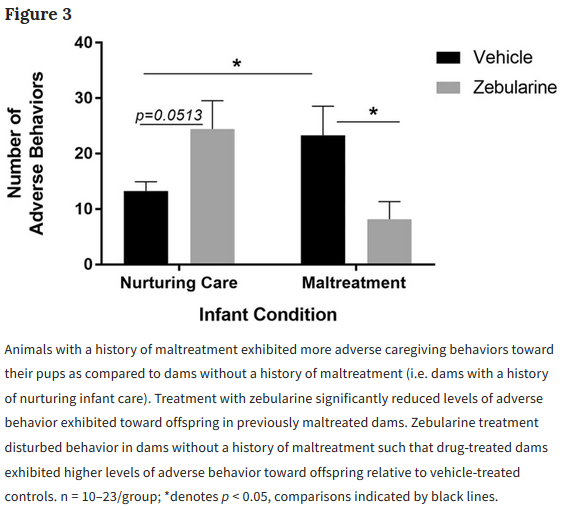
A pair of 2019 Virginia studies involved human mother/infant subjects:
“We show that OXTRm [oxytocin receptor gene DNA methylation] in infancy and its change is predicted by maternal engagement and reflective of behavioral temperament.”
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6795517 “Epigenetic dynamics in infancy and the impact of maternal engagement”
“Infants with higher OXTRm show enhanced responses to anger and fear and attenuated responses to happiness in right inferior frontal cortex, a region implicated in emotion processing through action-perception coupling.
Infant fNIRS [functional near-infrared spectroscopy] is limited to measuring responses from cerebral cortex. It is unknown whether OXTR is expressed in the cerebral cortex during prenatal and early postnatal human brain development.”

https://www.sciencedirect.com/science/article/pii/S187892931830207X “Epigenetic modification of the oxytocin receptor gene is associated with emotion processing in the infant brain”
Both studies had weak disclosures of limitations on their findings’ relevance and significance. The largest non-disclosed contrary finding was from the 2015 Early-life epigenetic regulation of the oxytocin receptor gene:
These results suggest that:
- Blood Oxtr DNA methylation may reflect early experience of maternal care, and
- Oxtr methylation across tissues is highly concordant for specific CpGs, but
- Inferences across tissues are not supported for individual variation in Oxtr methylation.
That rat study found that blood OXTR methylation of 25 CpG sites couldn’t accurately predict the same 25 CpG sites’ OXTR methylation in each subject’s hippocampus, hypothalamus, and striatum (which includes the nucleus accumbens) brain areas. Without significant effects in these limbic system structures, there couldn’t be any associated behavioral effects.
But CpG site associations and correlations were deemed good in the two current studies because they cited:
“Recent work in prairie voles has found that both brain- and blood-derived OXTRm levels at these sites are negatively associated with gene expression in the brain and highly correlated with each other.”
https://www.sciencedirect.com/science/article/pii/S0306453018306103 “Early nurture epigenetically tunes the oxytocin receptor”
The 2018 prairie vole study – which included several of the same researchers as the two current studies – found four nucleus accumbens CpG sites that had high correlations to humans. Discarding one of these CpG sites allowed their statistics package to make a four-decimal place finding:
“The methylation state of the blood was also associated with the level of transcription in the brain at three of the four CpG sites..whole blood was capable of explaining 94.92% of the variance in Oxtr DNA methylation and 18.20% of the variance in Oxtr expression.”
Few limitations on the prairie vole study findings were disclosed. Like the two current studies, there wasn’t a limitation section that placed research findings into suitable contexts. So readers didn’t know researcher viewpoints on items such as:
- What additional information showed that 3 of the 30+ million human CpGs accurately predicted specific brain OXTR methylation and expression from saliva OXTR methylation?
- What additional information demonstrated how “measuring responses from cerebral cortex” although “it is unknown whether OXTR is expressed in the cerebral cortex” provided detailed and dependable estimates of limbic system CpG site OXTR methylation and expression?
- Was the above 25-CpG study evidence considered?
Further contrast these three studies with a typical, four-point, 285-word limitation section of a study like Prenatal stress heightened adult chronic pain. The word “limit” appeared 6 times in that pain study, 3 times in the current fNIRS study, and 0 times in the current maternal engagement and cited prairie vole studies.
Frank interpretations of one’s own study findings to acknowledge limitations is one way researchers can address items upfront that will be questioned anyway. Such analyses also indicate a goal to advance science.