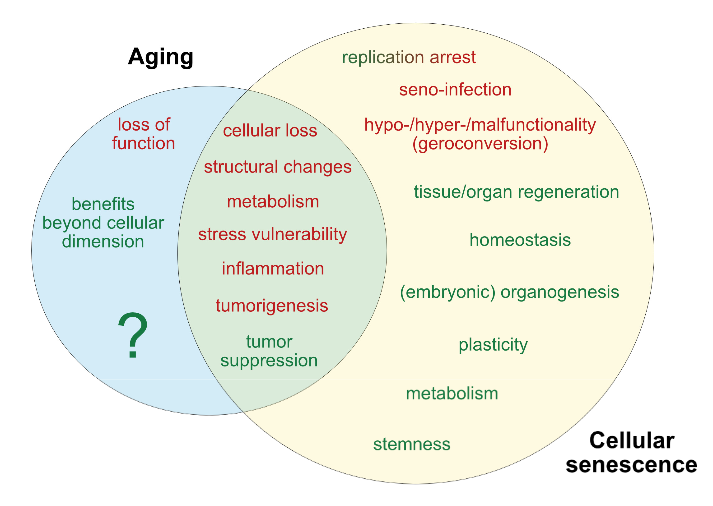
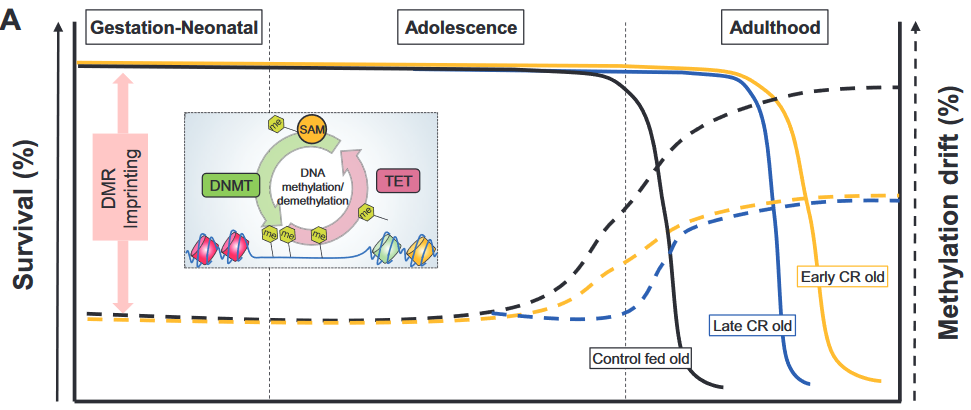
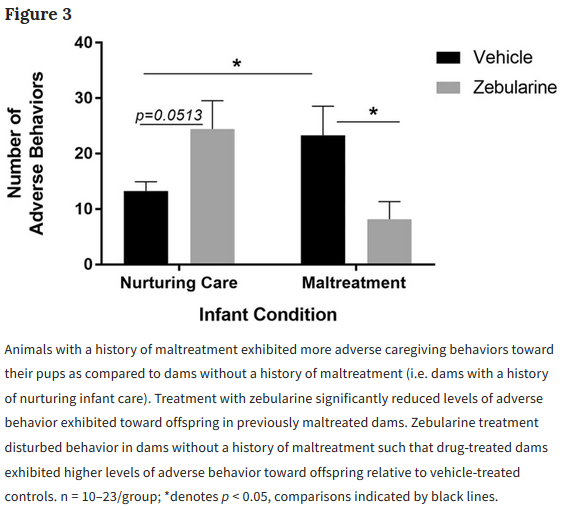
This 2020 review by a Hong Kong company’s researchers compared and contrasted measures of biological age:
“More than a dozen aging clocks use molecular features to predict an organism’s age, each of them utilizing different data types and training procedures. We offer a detailed comparison of existing mouse and human aging clocks, discuss their technological limitations and the underlying machine learning algorithms. We also discuss promising future directions of research.

Biomarkers placed on an intuitive plane of Accuracy vs Utility. Bubble size depends on the number of clocks based on a corresponding aging biomarker.
Currently, DNAm [DNA methylation] is the most accurate and the most frequently used biomarker in biohorology. However, it is harder to apply a DNAm clock compared to clocks based on clinical blood tests. Moreover, DNAm marks often take a long time to emerge in response to aging interventions.
Chromatin structure and telomeres, while intriguing, are too labor intensive and error-prone to be practical.”
https://www.sciencedirect.com/science/article/pii/S1568163719302582 “Biohorology and biomarkers of aging: current state-of-the-art, challenges and opportunities”
We think about chronological age linearly. The reviewers hinted at but didn’t directly assess the extent to which techniques such as linear regression may also influence people to think linearly about biological age.
We experience cyclical changes every day (like sleep), month, season, and longer periods. The reviewers didn’t mention techniques that incorporate our cyclical experiences or assess cyclical biological age.
1. The reviewers pointed out some biological age clock linearity flaws:
“Most aging clocks base their BA [biological age] definitions either on CA [chronological age] or mortality risk. Mortality risk in its turn is derived from demographic tables and can be assumed to be a function of CA in most animals, including human.
Thus, aging clocks are ultimately treating CA as a substitute BA with the caveat that deviations from the actual CA signify better or worse physical fitness when compared to age matched controls. Such a design has several flaws.”
2. They pointed out non-linear characteristics of chromosomal telomere length:
“DNA lesions caused by oxidative stress are repaired less efficiently in telomeric regions, which causes frailty and subsequent telomere shortening. Oxidative stress levels may fluctuate due to habitat, life style, inflammatory diseases – factors that do not necessarily represent replicative clock ticking.
Telomere length typically fluctuates within ±2-4% per month. This led scientists to hypothesize that telomere attrition is an oscillatory process.”
Since cell components show cyclical phases, why wouldn’t cells and each higher living structural level likewise demonstrate cyclical phases? That avenue wasn’t explored.
3. They mentioned the non-linearity of epigenetic clocks:
“If an organism’s DNAm profile is not directly linked to the thermodynamic root of aging [entropy] but instead is a downstream product of competing processes, the applicability of DNAm aging clock methodology is at risk. In this case different aging clocks may not be equally good for different experiment settings.
While genetic, pharmacological and dietary interventions with proven effect on life expectancy change the methylation state of the age-associated CpG sites, they do so in different ways. Caloric restriction is more efficient in preventing methylation loss at hypomethylated sites and methylation gain at hypermethylated sites than rapamycin.
These findings imply that DNAm profiles do not simply gravitate towards the average with age and that there is no single pathway through which all aging processes are imbued into an organism’s epigenetic landscape.”
4. Genetic and epigenetic regulatory pathways were presented with linear thinking:
“Protein structures encapsulating DNA and regulating its accessibility (chromatin and histones) have also been shown to change with age. Moreover, DNAm machinery and histone modifications are interlinked and change throughout aging concordantly.
For example, DNA methyltransferases are attracted by the H3K36me mark. With aging it is less tightly regulated, and thus, more sporadic DNAm occurs, which ultimately translates to epigenetic clock ticking.”
An individual’s capability to regulate their own aging phenotype wasn’t addressed, only externally applied “aging interventions.” Diseases were considered chronological-“age-associated.”
Biological aging was neither viewed as a disease nor as an unintended consequence. If these researchers don’t grasp the foundations of their field of study, why do they work in the biological aging field? It isn’t just math.
- Could this paper reflect one company’s desire to frame arguments in favor of the company’s offered solution?
- Could this paper reflect a “chronological age is the cause” meme that satisfied organizational imperatives for sponsors like the Buck Institute for Research on Aging?
- Or could it be that the reviewers had other paradigms?
What do you think?