Continuing Plasmalogens Week with two 2025 papers, starting with a rodent study of plasmalogens’ effects on mitigating cognitive decline:
“We evaluated beneficial effects of plasmalogens (PLS), phosphatidylcholine (PC), and phosphatidylserine (PS) on age-associated cognitive decline. We established a mouse model of aging-associated cognitive impairment using the subcutaneous injection of d-galactose (D-gal) at a dosage of 400 mg/kg/day.
We randomly divided six-week-old female mice into nine groups: control, model, high-dose PLS (0.3 mg/kg/day), low-dose PLS (0.09 mg/kg/day), high-dose PC (200 mg/kg/day), low-dose PC (50 mg/kg/day), high-dose PS (200 mg/kg/day), low-dose PS (50 mg/kg/day), AMC-Plas (120 mg/kg/day; and functional component PLS (0.252 mg/kg/day).
We administered PLS, PC, and PS separately by oral gavage once daily. We extracted PLS from scallops according to the literature. AMC-Plas is a commercially available health supplement known for its neuroprotective properties and memory-enhancing effects. In this study, we included AMC-Plas as a positive control group to evaluate the effects of different phospholipids.

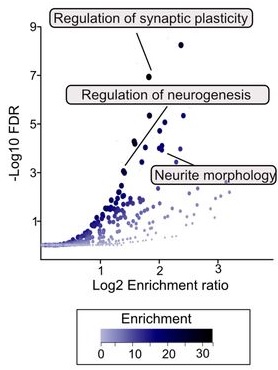
Synaptophysin (SYP), synapsin-1 (SYN-1), postsynaptic density protein 95 (PSD-95), and brain-derived neurotrophic factor (BDNF) play important roles in synapse formation and synaptic plasticity. Synaptic function alterations or losses are key pathological mechanisms that underlie development of cognitive impairment. Therapeutic strategies that attempt to restore synaptic function or promote synaptic remodeling are considered to be increasingly promising strategies to mitigate cognitive decline.
Results showed that:
- PLS improved spatial memory performance by 44% and object recognition by 80% in D-galactose-induced cognitively impaired mice.
- PLS significantly decreased glial fibrillary acidic protein (GFAP)-positive cells (an indicator of astrocyte activation) in the dentate gyrus (DG) of the hippocampus, an important result because the DG is a crucial neurogenesis region.
- PLS alleviated neuronal damage and protected against synaptic injury, verified by a 228% increase in PSD-95 expression in the hippocampus.
- PLS showed a more prominent role for the mitigation of age-related cognitive impairment compared with PC and PS.
In conclusion, the evaluation of PLS using both behavioral and neuropathological assessments in cognitively impaired mice highlighted its exceptional efficacy compared with other phospholipids. PLS at a remarkably low effective dose significantly ameliorated cognitive deficits in cognitively impaired mice. This result further emphasized its potential relevance in neurodegenerative disease research.
We found that PLS alleviated cognitive impairment potentially by improving synaptic function; however, the molecular mechanisms that underlie its effects on synaptic function warrant further investigation.”
https://www.sciencedirect.com/science/article/pii/S175646462500132X “Mitigating effects of plasmalogens on age-related cognitive impairment”
There was no disclosed chemical analysis of the PLS scallop extract’s plasmalogen types or other contents. Despite its name, I didn’t see that the AMC-Plas product contained plasmalogens or plasmalogen precursors.
A fruit fly study investigated plasmalogen effects on mitochondria during aging:
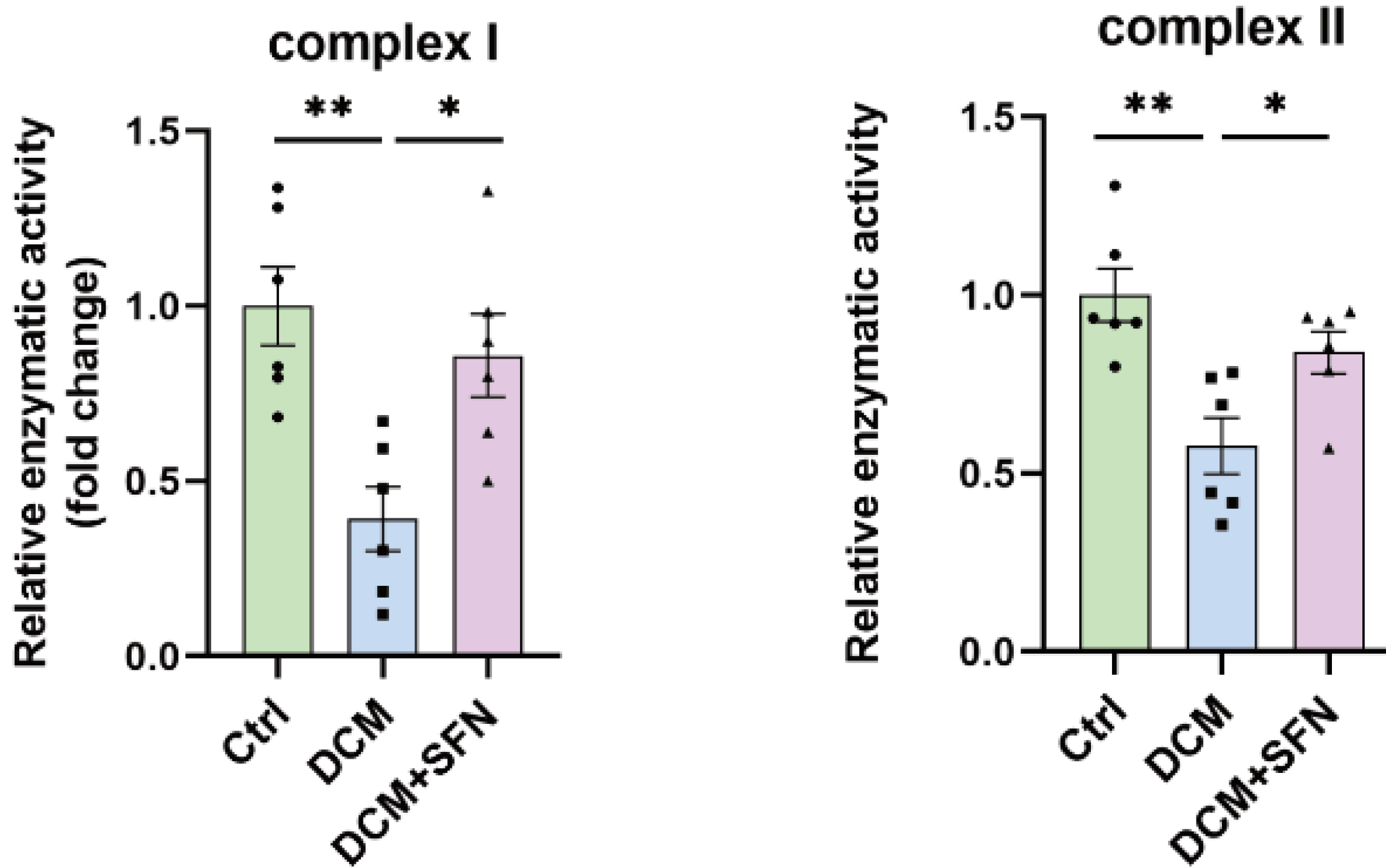
“We identify plasmalogens—endogenous ether-linked phospholipids—as key regulators of age-associated mitochondrial fission in Drosophila melanogaster. Loss of Kua (also known as plasmanylethanolamine desaturase (PEDS) / TMEM189 in mammals), the enzyme essential for plasmalogen biosynthesis, leads to inhibition of mitochondrial fission and impaired recruitment of the fission protein Drp1, similar to what is observed during aging.
Mitochondrial dynamics, comprising balanced cycles of fission and fusion, are essential for preserving organelle quality, metabolic flexibility, and cellular homeostasis throughout life. Aging disrupts this balance, with multiple studies reporting a decline in mitochondrial fission that contributes to the accumulation of enlarged and dysfunctional mitochondria.
These morphological changes are linked to impaired mitophagy, altered energy production, and tissue dysfunction. Midlife induction of Drp1—the dynamin-related GTPase that drives mitochondrial division—has been shown to reverse age-related mitochondrial defects and prolong lifespan in Drosophila.
To determine whether plasmalogen biosynthesis is essential for mitochondrial fission, we used KuaMI04999, a hypomorphic allele. Western blot analysis revealed significantly reduced Kua protein levels in KuaMI04999/+ heterozygotes compared to wild-type controls.

Our findings reveal a previously unrecognized lipid-based mechanism that controls mitochondrial fission during aging and position plasmalogens as key effectors linking membrane composition to mitochondrial homeostasis. It is not merely expression or stability of Drp1 that is affected, but rather its recruitment to the mitochondrial surface, which is a critical activation step for fission.
While our study highlights the requirement of plasmalogen biosynthesis for Drp1 recruitment, further work is needed to understand how plasmalogens mechanistically facilitate this interaction.”
https://www.researchsquare.com/article/rs-7330024/v1 “Plasmalogen Biosynthesis Controls Mitochondrial Fission via Drp1 Recruitment during Aging”
This study didn’t analyze or characterize specific plasmalogens.