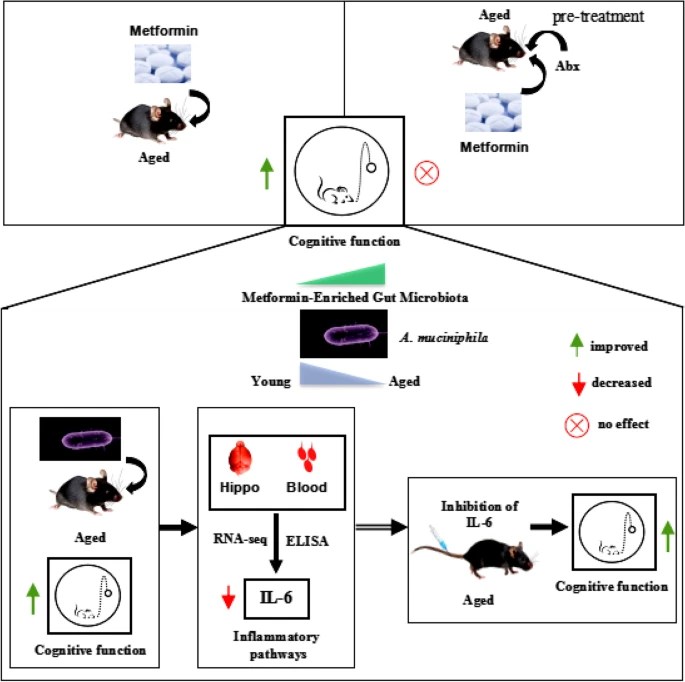
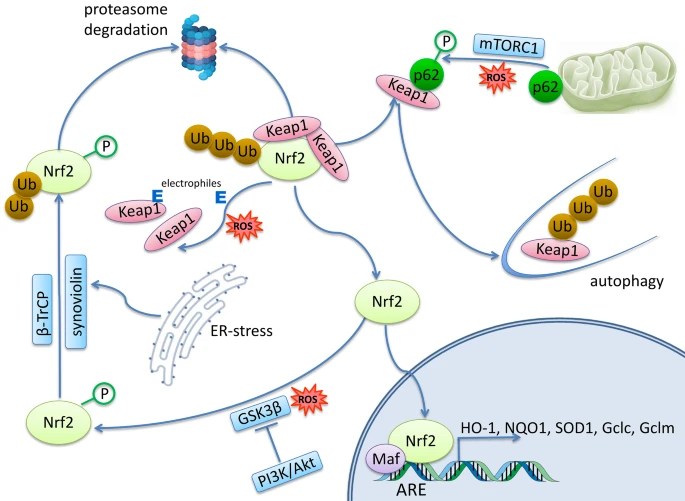
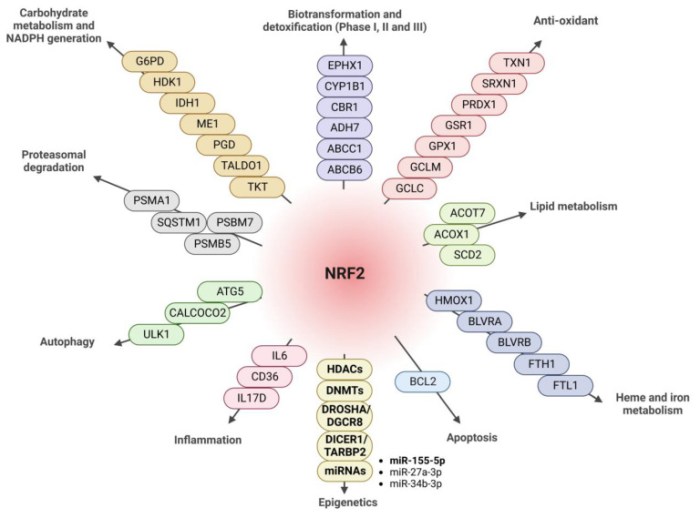
Six 2023 papers on the subject, starting with a rodent study:
“One of the primary discoveries of our study is that the endothelial cell (EC) transcriptome is dynamically regulated by both aging and heterochronic parabiosis. We found that ECs, when compared with other brain cell types, exhibited one of the highest fractions of aging-related genes that were rescued after heterochronic parabiosis in the old brain, and similarly, the highest fraction of aging-related genes that were disrupted after heterochronic parabiosis in the young brain. This finding supports our previous research that vasculature is strongly affected by aging and disease, and is capable of regrowth after heterochronic parabiosis or systemic GDF11 treatment.

We observed that a subset of ECs was classified as mitogenic. It is reasonable to speculate that the growth of these cells, which is probably prevented or suspended by the inflammatory environment of the aged brain, may be among the cell populations that respond to these interventions.
Although proteostasis in brain ECs has not been thoroughly investigated, they are apparently long-lived cells and, like neurons, might therefore accumulate protein aggregates with age, potentially compromising their function. ECs become senescent with age, but parabiosis may reverse that phenotype as well.
These findings underline the strong susceptibility and malleability of ECs, which are directly exposed to secreted factors in both brain parenchyma and blood, to adapt to changes in their microenvironment. ECs, despite comprising <5% of the total number of brain cells, are a promising and accessible target for treatment of aging and its associated diseases.”
https://www.nature.com/articles/s43587-023-00373-6 “Heterochronic parabiosis reprograms the mouse brain transcriptome by shifting aging signatures in multiple cell types”
A review elaborated on endothelial cell senescence:
“ECs form highly dynamic and differentiated monolayers arranged in a vascular network. Within brain tissue, the ECs of arteries, capillaries, and veins present different molecular characteristics. The main functions of ECs as a major cellular component of the blood-brain barrier (BBB) are to express cell membrane transport proteins, produce inflammatory mediators, deliver nutrients to brain tissue, and prevent drugs and toxins from entering the central nervous system.
ECs are the first echelons of cells affected at the onset of senescence due to their special structural position in the vascular network. Senescent ECs produce reactive oxygen species (ROS), which directly inhibit smooth muscle potassium channels and cause vasoconstriction.
The vascular endothelium is in a constant process of damage and repair, and once damage occurs, ECs replenish themselves to remove damaged cells. EC senescence makes the endothelium less capable of self-repair. With the decline in endothelial function, excess accumulated senescent cells express senescence-associated secretory phenotypes (SASPs), which result in senescence of adjacent cells, and eventually degeneration of vascular function.”
https://www.aginganddisease.org/EN/10.14336/AD.2023.0226-1 “Endothelial Senescence in Neurological Diseases”
A human study investigated above-mentioned differences in brain endothelial cells:
“We performed single nucleus RNAseq on tissue from 32 Alzheimer’s Disease (AD) and non-AD donors each with five cortical regions: entorhinal cortex, inferior temporal gyrus, prefrontal cortex, visual association cortex, and primary visual cortex. Analysis of 51,586 endothelial cells revealed unique gene expression patterns across the five regions in non-AD donors.
Visual cortex areas, which are affected late in AD progression and experience less neurodegeneration, expressed more genes related to vasculogenesis and angiogenesis. Highly vulnerable areas such as the entorhinal cortex expressed more oxidative stress-related genes in normal aged brain, suggesting endothelial dysfunction in this region even in the absence of severe AD pathology.
The present work shows that senescence-related gene signatures are increased across several brain regions, and confirms these changes in endothelial cells in the absence of other vascular cell types. While endothelial cells are not typically associated with protein aggregation, upregulated protein folding pathways suggest that proteostatic stress is a key pathway in this cell type.”
https://www.biorxiv.org/content/10.1101/2023.02.16.528825v1.full “Endothelial Cells are Heterogeneous in Different Brain Regions and are Dramatically Altered in Alzheimer’s Disease”
A human cell study abstract on above-mentioned blood-brain barrier endothelial cells:
“The BBB is a semi-permeable and protective barrier of the brain, primarily composed of endothelial cells interconnected by tight junction proteins, that regulates movement of ions and molecules between blood and neural matter. In pathological conditions such as traumatic brain injury (TBI), disruption of the BBB contributes to leakage of solutes and fluids into brain parenchyma, resulting in onset of cerebral edema and elevation of intracranial pressure.
The objective of this study was to determine upstream regulators of NLRP3 signaling and BBB hyperpermeability, particularly to determine if extracellular adenosine triphosphate (ATP) via P2X7R, a purinergic receptor, promotes NLRP3 inflammasome activation. Extracellular ATP is a major contributor of secondary injuries following TBI.
Our results suggest that extracellular ATP promotes NLRP3 inflammasome activation. Subsequent caspase-1 and MMP-9-mediated tight junction disorganization are major pathways that lead to BBB dysfunction and hyperpermeability following conditions such as TBI.”
https://journals.physiology.org/doi/abs/10.1152/physiol.2023.38.S1.5732827 “Regulation of Blood-Brain Barrier Endothelial Cell Hyperpermeability by NLRP3 Inflammasome Inhibition”
A human study further investigated effects of traumatic brain injury on brain endothelial cells:
“We previously demonstrated that extracellular vesicles (EVs) released from injured brains led to endothelial barrier disruption and vascular leakage. Here, we enriched plasma EVs from TBI patients (TEVs), detected high mobility group box 1 (HMGB1) exposure to 50.33 ± 10.17% of TEVs, and found the number of HMGB1+TEVs correlated with injury severity. We then investigated for the first time the impact of TEVs on endothelial function using adoptive transfer models.
HMGB1 is secreted by activated cells or passively released by necrotic or injured cells. After post-translational modifications, it interacts with receptors such as toll-like receptors (TLRs; e.g., TLRs 2, 4, and 9) and the receptor for advanced glycation end products (RAGE) to trigger multiple signaling pathways and mediate inflammatory and immune responses. Extracellular HMGB1 promotes endothelial dysfunction, leukocyte activation and recruitment, as well as thrombosis.
These results suggest that circulating EVs isolated from patients with TBI alone are sufficient to induce endothelial dysfunction. They contribute to secondary brain injury that are dependent on immunologically active HMGB1 exposed on their surface. This finding provided new insight for development of potential therapeutic targets and diagnostic biomarkers for TBI.”
https://www.sciencedirect.com/science/article/pii/S1043661823001470 “Circulating extracellular vesicles from patients with traumatic brain injury induce cerebrovascular endothelial dysfunction”
To wrap up, eat mushrooms to protect your brain endothelial cells!
“Natural compound ergothioneine (ET), which is synthesised by certain fungi and bacteria, has considerable cytoprotective potential. We previously demonstrated anti-inflammatory effects of ET on 7-ketocholesterol (7KC)-induced endothelial injury in human blood-brain barrier endothelial cells (hCMEC/D3). 7KC is an oxidised form of cholesterol present in atheromatous plaques and sera of patients with hypercholesterolaemia and diabetes mellitus. The aim of this study was to elucidate the protective effect of ET on 7KC-induced mitochondrial damage.
Protective effects of ET were diminished when endothelial cells were coincubated with verapamil hydrochloride (VHCL), a nonspecific inhibitor of the ET transporter OCTN1 (SLC22A4). This outcome demonstrates that ET-mediated protection against 7KC-induced mitochondrial damage occurred intracellularly and not through direct interaction with 7KC.
OCTN1 mRNA expression itself was significantly increased in endothelial cells after 7KC treatment, consistent with the notion that stress and injury may increase ET uptake. Our results indicate that ET can protect against 7KC-induced mitochondrial injury in brain endothelial cells.”
https://www.mdpi.com/1422-0067/24/6/5498 “Protective Effect of Ergothioneine against 7-Ketocholesterol-Induced Mitochondrial Damage in hCMEC/D3 Human Brain Endothelial Cells”