Experiential feeling therapy addressing the pain of the lack of love.
Infants
A limited study of parental transmission of anxiety/stress-reactive traits
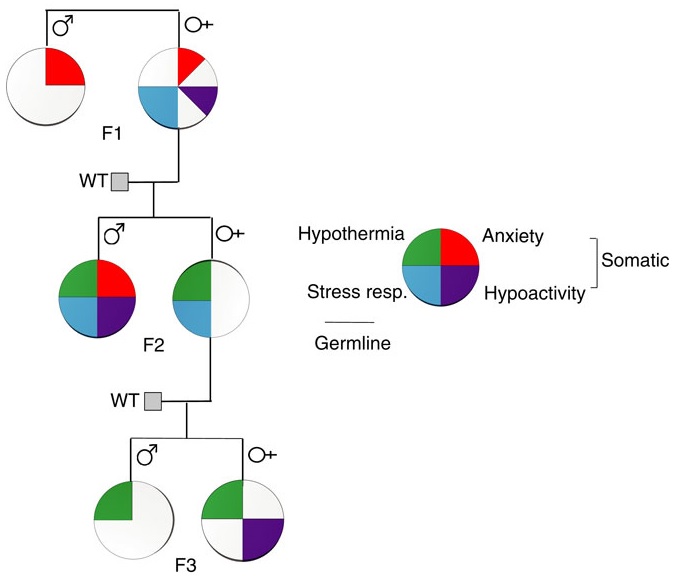 This 2016 New York rodent study found:
This 2016 New York rodent study found:
“Parental behavioural traits can be transmitted by non-genetic mechanisms to the offspring.
We show that four anxiety/stress-reactive traits are transmitted via independent iterative-somatic and gametic epigenetic mechanisms across multiple generations.
As the individual traits/pathways each have their own generation-dependent penetrance and gender specificity, the resulting cumulative phenotype is pleiotropic. In the context of genetic diseases, it is typically assumed that this phenomenon arises from individual differences in vulnerability to the various effects of the causative gene. However, the work presented here reveals that pleiotropy can be produced by the variable distribution and segregated transmission of behavioural traits.”
A primary focus was how anxiety was transmitted from parents to offspring:
“The iterative propagation of the male-specific anxiety-like behaviour is most compatible with a model in which proinflammatory state is propagated from H [serotonin1A receptor heterozygote] F0 to F1 [children] females and in which the proinflammatory state is acquired by F1 males from their H mothers, and then by F2 [grandchildren] males from their F1 mothers.
We propose that increased levels of gestational MIP-1β [macrophage inflammatory protein 1β] in H and F1 mothers, together with additional proinflammatory cytokines and bioactive proteins, are required to produce immune system activation in their newborn offspring, which in turn promotes the development of the anxiety-like phenotype in males.
In particular, increase in the number of monocytes and their transmigration to the brain parenchyma in F1 and F2 males could be central to the development of anxiety.”
The researchers studied transmission of behavioral traits and epigenetic changes. Due to my quick take on the study title – “Behavioural traits propagate across generations..” – I had expectations of this study that weren’t born out. What could the researchers have done versus what they did?
The study design removed prenatal and postnatal parental behavioral transmission of behavioral traits and epigenetic changes as each generation’s embryos were implanted into foster wild-type (WT) mothers.
The study design substituted the foster mothers’ prenatal and postnatal parental environments for the biological parents’ environments. So we didn’t find out, for example:
- To what extents the overly stress-reactive F1 female children’s prenatal environments and postnatal behaviors induced behaviors and/or epigenetic changes in their children; and
- Whether the F2 grandchildren’s parental behaviors subsequently induced behaviors and/or epigenetic changes in the F3 great-grandchildren.
How did the study meet the overall goal of rodent studies: to help humans?
-
- Only a minority of humans experienced an early-life environment that included primary caregivers other than our biological parents.
- Very, very few of us experienced a prenatal environment other than our biological mothers.
- The study’s thorough removal of parental behavior was an outstanding methodology to confirm by falsifiability whether parental behavior was both an intergenerational and transgenerational epigenetic inheritance mechanism.
- Maybe the researchers filled in some gaps in previous rodent studies, such as determining what is or isn’t a “true transgenerational mechanism.”
As an example of a rodent study that more closely approximated human conditions, the behavior of a mother whose DNA was epigenetically changed by stress induced the same epigenetic changes to her child’s DNA when her child was stressed per One way that mothers cause fear and emotional trauma in their infants:
“Our results provide clues to understanding transmission of specific fears across generations and its dependence upon maternal induction of pups’ stress response paired with the cue to induce amygdala-dependent learning plasticity.”
How did parental behavioral transmission of behavioral traits and epigenetic changes become a subject not worth investigating? These traits and effects can be seen everyday in real-life human interactions, and in every human’s physiology.
But when investigating human correlates with behavioral epigenetic changes of rodents in the laboratory, parental behavioral transmission of behavioral traits is often treated the way this study treated it: as a confounder.
I doubt that people who have reached some degree of honesty about their early lives and concomitant empathy for others would agree with this prioritization. The papers of Transgenerational epigenetic inheritance week show the spectrum of opportunities to advance science that were intentionally missed.
http://www.nature.com/ncomms/2016/160513/ncomms11492/full/ncomms11492.html “Behavioural traits propagate across generations via segregated iterative-somatic and gametic epigenetic mechanisms”
Does childhood trauma influence offspring’s birth characteristics?
This 2016 Swedish human study investigated the effects of one specific childhood trauma, parental death:
“Parental (G1) death during (G2) childhood predicts prematurity and lower birthweight in the offspring generation (G3). This response is dependent on G2 gender, G2 age at exposure and G3 parity, but not on G3 gender.
Offspring of women who lost their parent at the age of 0-2 or at the age of 13-17 had an increased risk for prematurity.
Offspring of men who lost a parent at ages 8-12 had an increased risk of prematurity.
For women exposed to a parent’s death at age 0-2, there was no significant deficit in their offspring’s birthweight in any parity class. For women exposed at later ages we observed a deficit in birthweight.
Among children whose fathers experienced parental loss..experiencing parental death at ages 8-12 in particular, or at ages 13-17, but not at ages 0-2 or 3-7, did predict having lighter offspring.”
The study design was unable to produce causal evidence for the putative intergenerational effects. An example of the limitations was:
“We had no information about behaviours and biological markers or genes.”
Its findings were best summarized as:
“Our study fails to refute the hypothesis that a male-line epigenetic mechanism exists which may be triggered by trauma during boys’ slow growth period.”
Still, the study had a firmer foundation than did A problematic study of oxytocin receptor gene methylation, childhood abuse, and psychiatric symptoms, which speciously produced politically-correct results from childhood trauma surveys of adults.
http://ije.oxfordjournals.org/content/early/2016/05/03/ije.dyw048.full “Does childhood trauma influence offspring’s birth characteristics?”
Using salivary microRNA to diagnose autism
This 2016 New York human study found:
“Measurement of salivary miRNA in this pilot study of subjects with mild ASD [autism spectrum disorder] demonstrated differential expression of 14 miRNAs that are:
Some problems with current diagnostic methods for autism are:
“The first sign of ASD commonly recognized by pediatricians is a deficit in communication and language that does not manifest until 18–24 months of age.
The mean age of diagnosis for children with ASD is 3 years, and approximately half of these are false-positives.
Despite a substantial genetic component, no single gene variant accounts for >1 % of ASD incidence.
Nearly 2000 individual genes have been implicated in ASD, but none are specific to the disorder.”
Study limitations included:
“Aside from the sample size and cross-sectional nature of this pilot study, another limitation is the age of ASD and control subjects it describes (4–14 years) which are not representative of the target population in which ASD biomarkers would ideally be utilized (0–2 years). However, selecting a homogenous group of subjects with mild ASD (as measured by ADOS) that was well-established and diagnosed by a developmental specialist requires subjects with long-standing diagnoses.”
Understanding later-life consequences of disrupted neurodevelopment is critical for tracing symptoms back to their causes, as noted in Grokking an Adverse Childhood Experiences (ACE) score. I wonder how long it will take for researchers in other fields to stop wasting resources and do what this study did: focus on epigenetic biomarkers that have developmental origins.
http://bmcpediatr.biomedcentral.com/articles/10.1186/s12887-016-0586-x “Salivary miRNA profiles identify children with autism spectrum disorder, correlate with adaptive behavior, and implicate ASD candidate genes involved in neurodevelopment”
A one-sided review of stress
The subject of this 2016 Italian/New York review was the stress response:
“The stress response, involving the activation of the hypothalamic-pituitary-adrenocortical [HPA] axis and the consequent release of corticosteroid hormones, is indeed aimed at promoting metabolic, functional, and behavioral adaptations. However, behavioral stress is also associated with fast and long-lasting neurochemical, structural, and behavioral changes, leading to long-term remodeling of glutamate transmission, and increased susceptibility to neuropsychiatric disorders.
Of note, early-life events, both in utero and during the early postnatal life, trigger reprogramming of the stress response, which is often associated with loss of stress resilience and ensuing neurobehavioral (mal)adaptations.”
The reviewers’ intentional dismissal of the role of GABA in favor of the role of glutamate was a key point:
“The changes in neuronal excitability and synaptic plasticity induced by stress are the result of an imbalance of excitatory (glutamatergic) and inhibitory (GABAergic) transmission, leading to long-lasting (mal)adaptive functional modifications. Although both glutamate and GABA transmission are critically associated with stress-induced alteration of neuronal excitability, the present review will focus on the modulation of glutamate release and transmission induced by stress and glucocorticoids.”
No particular reason was given for this bias. I inferred from the review’s final sentence that the review’s sponsors and funding prompted this decision:
“In-depth studies of changes in glutamate transmission and dendrite remodeling induced by stress in early and late life will help to elucidate the biological underpinnings of the (mal)adaptive strategies the brain adopts to cope with environmental challenges in one’s life.”
The bias led to ignoring evidence for areas the reviewers posed as needing further research. An example of relevant research the reviewers failed to consider was the 2015 Northwestern University study I curated in A study that provided evidence for basic principles of Primal Therapy that found:
“In response to traumatic stress, some individuals, instead of activating the glutamate system to store memories, activate the extra-synaptic GABA system and form inaccessible traumatic memories.”
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4812483/ “Stress Response and Perinatal Reprogramming: Unraveling (Mal)adaptive Strategies”
The current paradigm of child abuse limits pre-childhood causal research
As an adult, what would be your primary concern if you suspected that your early life had something to do with current problems? Would you be interested in effective treatments for causes of your symptoms?
Such information wasn’t available in this 2016 Miami review of the effects of child abuse. The review laid out the current paradigm mentioned in Grokking an Adverse Childhood Experiences (ACE) score, one that limits research into pre-childhood causes for later-life symptoms.
The review’s goal was to describe:
“How numerous clinical and basic studies have contributed to establish the now widely accepted idea that adverse early life experiences can elicit profound effects on the development and function of the nervous system.”
The hidden assumptions of almost all of the cited references were that these distant causes could no longer be addressed. Aren’t such assumptions testable today?
As an example, the Discussion section posed the top nine “most pressing unanswered questions related to the neurobiological effects of early life trauma.” In line with the current paradigm, the reviewer assigned “Are the biological consequences of ELS [early life stress] reversible?” into the sixth position.
If the current paradigm encouraged research into treatment of causes, there would probably already be plenty of evidence to demonstrate that directly reducing the source of damage would also reverse damaging effects. There would have been enough studies done so that the generalized question of reversibility wouldn’t be asked.
Aren’t people interested in treatments of originating causes so that their various symptoms don’t keep bubbling up? Why wouldn’t research paradigms be aligned accordingly?
The review also demonstrated how the current paradigm of child abuse misrepresented items like telomere length and oxytocin. Researchers on the bandwagon tend to forget about the principle Einstein expressed as:
“No amount of experimentation can ever prove me right; a single experiment can prove me wrong.”
That single experiment for telomere length arrived in 2016 with Using an epigenetic clock to distinguish cellular aging from senescence. The review’s seven citations for telomere length that all had findings “associated with” or “linked to” child abuse should now be viewed in a different light.
The same light shone on oxytocin with Testing the null hypothesis of oxytocin’s effects in humans and Oxytocin research null findings come out of the file drawer. See their references, and decide for yourself whether or not:
“Claimed research findings may often be simply accurate measures of the prevailing bias.”
http://www.cell.com/neuron/fulltext/S0896-6273%2816%2900020-9 “Paradise Lost: The Neurobiological and Clinical Consequences of Child Abuse and Neglect”
This post has somehow become a target for spammers, and I’ve disabled comments. Readers can comment on other posts and indicate that they want their comment to apply here, and I’ll re-enable comments.
Beneficial epigenetic effects of mild stress with social support during puberty
This 2016 Pennsylvania rodent study found:
“Stress in the context of social support experienced over the pubertal window can promote epigenetic reprogramming in the brain to increase resilience to age-related cognitive decline in females.
These findings are actually consistent with previous studies showing that some amount of adversity, or adversity under more favorable circumstances such as social support or a protective gene polymorphism, provides a measure of ‘grit’ in coping with later life challenges.
Our findings provide a unique perspective on this relationship, as they highlight the important link between experience during the pubertal window and cognitive health during aging.”
These researchers made efforts to further investigate causes of unexpected results, such as:
“Peripubertal stress alone did not significantly alter Barnes maze performance in aging compared to aged Controls. Mice that had experienced stress with concurrent social support (CVS + SI) actually performed better than Control aged mice, specifically in learning the reversal task faster.
Peripubertal stress had no effect on corticosterone levels in response to an acute restraint stress or in sensorimotor gating and baseline startle reactivity.”
Their investigations led to epigenetic findings:
“Consistent with our behavioral findings, stress in the context of social interaction resulted in long-term reprogramming of gene expression in the PFC [prefrontal cortex]. While there were no differentially expressed genes between Control and CVS females, there were 88 genes that were significantly different between Control and CVS + SI groups. Of genes that were downregulated, a large portion (23 genes; 35%) were microRNAs.
We found that the PFC transcriptome of CVS + SI aged females was significantly enriched for predicted targets of the 23 microRNAs that were downregulated in the PFC in these mice. This suggests that microRNAs represent a mode of regulation capable of enacting far-reaching programmatic effects, and are a critical epigenetic gene expression regulatory mechanism.”
Applicability to humans was suggested by associations such as:
“A single microRNA can target more than a hundred different mRNA targets, and more than 45,000 conserved microRNA binding sites have been annotated in the 3′ UTR of 60% of human genes.”
A few limitations were noted:
“Given that mice at this age (1 year) are commonly compared to ‘late middle aged’ humans, later aging time points may yield differences in this group. Alternatively, it is possible that there was an effect of peripubertal stress that was not long-lasting due to the mild nature of our chronic stress model.
To include early neglect as a part of the stressor experience, CVS females were weaned one week earlier (PN21) than Control and CVS + SI mice. Addition of stress of this earlier weaning likely poses a significant contribution to programming of the PFC.”
One of the study coauthors was also a coauthor of:
- Transgenerational epigenetic programming with stress and microRNA
- How to make a child less capable even before they are born: stress the pregnant mother-to-be
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4870871/ “Peripubertal stress with social support promotes resilience in the face of aging”
Using an epigenetic clock with children
This 2015 UK human study by many of the coauthors of What’s the origin of the problem of being fat? applied the Horvath epigenetic clock method to the same UK mother-child pairs and a Danish cohort:
“There has been no investigation on prenatal and antenatal factors that affect AA [age acceleration] in children. It is possible that the detrimental consequences of a higher AA may accrue over time, initiating in childhood. Conversely, it could be postulated that having a positive AA during early life and childhood is developmentally advantageous. To reflect this, we could refer to AA as an epigenetic measure of development in children.
We found associations between AA and sex, birth weight, caesarean section delivery and several maternal characteristics, namely smoking in pregnancy, weight, BMI, selenium and cholesterol level.
Offspring of non-drinkers had higher AA on average at birth, but this appeared to resolve during childhood. Offspring of smokers had higher AA on average and this difference became larger during childhood and adolescence.
The lack of correlation between AA and several clinical variables may also indicate that AA reflects an ‘intrinsic’ aging rate that is independent of various aging factors.
The observation that the estimated genetic component of AA increased in older study participants may indicate that the AA measure is more biologically meaningful in adults rather than children, though alternatively it could be a reflection of a decreasing environmental influence on DNA methylation patterns over time.
This accords with our finding of strengthening within subject correlation over time, which suggests the period of rapid early life changes in methylation affects epigenetic age during development to a greater extent than adulthood changes in methylation.”
The heritability of age acceleration was analyzed:
“The heritability estimate from our study (h = 0.37) is lower than that reported Horvath (h = 1.0), which was based on a small number of cord blood samples from twin pairs. Both of these heritability estimates were based on relatively few samples. Future large scale studies will be needed to arrive at precise estimates of the heritability of AA in newborns and minors.
While our heritability estimate may seem low, empirical evidence has suggested that fitness related traits tend to have lower heritability than morphological traits because selection acts to purify deleterious genetic variation, and one might consider age accelerated residuals in the former category.”
Like the coauthors’ follow-on study, causality couldn’t be definitively determined:
“Assessing the causal relationship between exposures and AA (through Mendelian randomization) is underpowered in our current data.”
Epigenetic age acceleration at birth seemed to be overall “developmentally advantageous” for offspring of non-drinking mothers. That age acceleration continued for the offspring of smokers at the second and third measurement times (ages 7 and 15-17) seemed to have “detrimental consequences.” I’d guess that the methylation state of specific CpG sites would be more informative than the overall rate in these cases.
The point about “AA..is independent of various aging factors” was similar to one made in Using an epigenetic clock to distinguish cellular aging from senescence:
“Cellular ageing is distinct from cellular senescence and independent of DNA damage response and telomere length.”
The study was a step toward establishing the Horvath epigenetic clock for widespread usage. The Hannum method was also compared and contrasted.
http://hmg.oxfordjournals.org/content/25/1/191.full “Prenatal and early life influences on epigenetic age in children: a study of mother-offspring pairs from two cohort studies”
What’s the origin of the problem of being fat?
This 2016 UK human study attempted to replicate the DNA methylation and adiposity associations found by studies on a long-term longitudinal UK cohort:
“We tested for replication of associations between previously identified CpG sites at HIF3A [the hypoxia inducible factor 3 alpha subunit gene] and adiposity in ∼1,000 mother-offspring pairs from the Avon Longitudinal Study of Parents and Children.”
The researchers had sufficient data to test the unidirectional and causal findings of previous studies:
“Availability of methylation and adiposity measures at multiple time points, as well as genetic data, allowed us to assess the temporal associations between adiposity and methylation and to make inferences regarding causality and directionality.”
The analyses didn’t replicate the previous studies’ findings, and a new finding was indicated:
“Our results were discordant with those expected if HIF3A methylation has a causal effect on BMI [body mass index, derived from height and weight] and provided more evidence for causality in the reverse direction i.e. an effect of BMI on HIF3A methylation.
These results are based on robust evidence from longitudinal analyses and were also partially supported by Mendelian randomization analysis, although this latter analysis was underpowered to detect a causal effect of BMI on HIF3A methylation.
Our results also highlight an apparent long-lasting inter-generational influence of maternal BMI on offspring methylation at this locus, which may confound associations between own [offspring] adiposity and HIF3A methylation.”
A person’s parents contributed all of their genetic material and the prenatal environment, and usually almost all of their postnatal and childhood development environment. If a person has a health problem that may have genetic and developmental origins, this is where to look for causes and preventive actions.
That these distant causes can no longer be addressed is a hidden assumption of research and treatment of effects of health problems. Aren’t such assumptions testable here in the current year?
http://diabetes.diabetesjournals.org/content/early/2016/02/01/db15-0996.long (pdf) “DNA methylation and body mass index: investigating identified methylation sites at HIF3A in a causal framework”
A problematic study of oxytocin receptor gene methylation, childhood abuse, and psychiatric symptoms
This 2016 Georgia human study found:
“A role for OXTR [oxytocin receptor gene] in understanding the influence of early environments on adult psychiatric symptoms.
Data on 18 OXTR CpG sites, 44 single nucleotide polymorphisms, childhood abuse, and adult depression and anxiety symptoms were assessed in 393 African American adults. The Childhood Trauma Questionnaire (CTQ), a retrospective self-report inventory, was used to assess physical, sexual, and emotional abuse during childhood.
While OXTR CpG methylation did not serve as a mediator to psychiatric symptoms, we did find that it served as a moderator for abuse and psychiatric symptoms.”
From the Limitations section:
- “Additional insight will likely be gained by including a more detailed assessment of abuse timing and type on the development of biological changes and adverse outcomes.
- The degree to which methylation remains fixed following sensitive developmental time periods, or continues to change in response to the environment, is still a topic of debate and is not fully known.
- Comparability between previous findings and our study is limited given different areas covered.
- Our study was limited to utilizing peripheral tissue [blood]. OXTR methylation should ideally be assessed in the tissues that are known to express OXTR and directly involved in psychiatric symptoms. The degree to which methylation of peripheral tissues can be used to study methylation changes in response to the environment or in association with behavioral outcomes is currently a topic of debate.
- Our study did not evaluate gene expression and thus cannot explore the role of study CpG sites on regulation and expression.”
Addressing the study’s limitations:
- Early-life epigenetic regulation of the oxytocin receptor gene demonstrated – with no hint of abuse – how sensitive an infant’s experience-dependent oxytocin receptor gene DNA methylation was to maternal care. Treating prenatal stress-related disorders with an oxytocin receptor agonist provided evidence for prenatal oxytocin receptor gene epigenetic changes.
- No human’s answers to the CTQ, Adverse Childhood Experiences, or other questionnaires will ever be accurate self-reports of their prenatal, infancy, and early childhood experiences. These early development periods were likely when the majority of the subjects’ oxytocin receptor gene DNA methylation took place. The CTQ self-reports were – at best – evidence of experiences at later times and places, distinct from earlier experience-dependent epigenetic changes.
- As one example of incomparability, the 2009 Genomic and epigenetic evidence for oxytocin receptor deficiency in autism was cited in the Introduction section and again in the Limitations section item 4. Since that study was sufficiently relevant to be used as a reference twice, the researchers needed to provide a map between its findings and the current study.
- Early-life epigenetic regulation of the oxytocin receptor gene answered the question of whether or not an individual’s blood could be used to make inferences about their brain oxytocin receptor gene DNA methylation. The evidence said: NO, it couldn’t.
- It’s assumed that oxytocin receptor gene DNA methylation directly impacted gene expression such that increased levels of methylation were associated with decreased gene transcription. The study assumed but didn’t provide evidence that higher levels of methylation indicated decreased ability to use available oxytocin due to decreased receptor expression. The study also had no control group.
To summarize the study’s limitations:
- The study zeroed in on childhood abuse, and disregarded evidence for more relevant factors determining an individual’s experience-dependent oxytocin receptor gene DNA methylation. That smelled like an agenda.
- The study used CTQ answers as determinants, although what happened during the subjects’ earliest life was likely when the majority of epigenetic changes to the oxytocin receptor gene took place. If links existed between the subjects’ early-life DNA methylation and later-life conditions, they weren’t evidenced by CTQ answers about later life that couldn’t self-report relevant experiences from conception through age three that may have caused DNA methylation.
- There was no attempt to make findings comparable with cited studies. That practice and the lack of a control group reminded me of Problematic research with telomere length.
- The researchers tortured numbers until they confessed “that CpG methylation may interact with abuse to predict psychiatric symptoms.” But there was no direct evidence that each subject’s blood oxytocin gene receptor DNA methylation interacted as such! Did the “may interact” phrase make the unevidenced inferences more plausible, or permit contrary evidence to be disregarded?
- See Testing the null hypothesis of oxytocin’s effects in humans for examples of what happens when researchers compound assumptions and unevidenced inferences.
The study’s institution, Emory University, and one of the study’s authors also conducted Conclusions without evidence regarding emotional memories. That 2015 study similarly disregarded relevant evidence from other research, and made statements that weren’t supported by that study’s evidence.
The current study used “a topic of debate” and other disclaimers to provide cover for unconvincing methods and analyses in pursuit of..what? What overriding goals were achieved? Who did the study really help?
http://onlinelibrary.wiley.com/enhanced/doi/10.1111/cdev.12493/ “Oxytocin Receptor Genetic and Epigenetic Variations: Association With Child Abuse and Adult Psychiatric Symptoms”
This post has somehow become a target for spammers, and I’ve disabled comments. Readers can comment on other posts and indicate that they want their comment to apply here, and I’ll re-enable comments.
Early-life epigenetic regulation of the oxytocin receptor gene
This 2015 US/Canadian rodent study investigated the effects of natural variation in maternal care:
“The effects of early life rearing experience via natural variation in maternal licking and grooming during the first week of life on behavior, physiology, gene expression, and epigenetic regulation of Oxtr [oxytocin receptor gene] across blood and brain tissues (mononucleocytes, hippocampus, striatum, and hypothalamus).
Rats reared by high licking-grooming (HL) and low licking-grooming (LL) rat dams exhibited differences across study outcomes:
- LL offspring were more active in behavioral arenas,
- Exhibited lower body mass in adulthood, and
- Showed reduced corticosterone responsivity to a stressor.
Oxtr DNA methylation was significantly lower at multiple CpGs in the blood of LL versus HL males, but no differences were found in the brain. Across groups, Oxtr transcript levels in the hypothalamus were associated with reduced corticosterone secretion in response to stress, congruent with the role of oxytocin signaling in this region.
Methylation of specific CpGs at a high or low level was consistent across tissues, especially within the brain. However, individual variation in DNA methylation relative to these global patterns was not consistent across tissues.
These results suggest that:
- Blood Oxtr DNA methylation may reflect early experience of maternal care, and
- Oxtr methylation across tissues is highly concordant for specific CpGs, but
- Inferences across tissues are not supported for individual variation in Oxtr methylation.
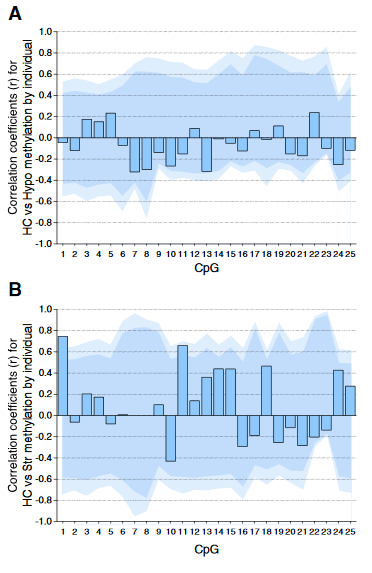
Individual DNA methylation values were not correlated across brain tissues, despite tissue concordance at the group level.
For each CpG, we computed the Pearson correlation coefficient r between methylation values for matched samples in pairs of brain regions (bars). Dark and light shaded regions represent 95% and 99% thresholds, respectively, of distributions of possible correlation coefficients determined from 10,000 permutations of the measured values among the individuals. These distributions represent the null hypothesis that an individual DNA methylation value in one brain region does not help to predict the value in another region in the same animal.
(A) Correlations based on pyrosequencing data for matched samples passing validation in both hippocampus (HC) and hypothalamus (Hypo). Correlations for individuals at each CpG were either weak (.2 < r < .3) or absent (r < .2), and none were significant, even prior to correction for multiple comparisons.
(B) Correlations for matched samples passing validation in both hippocampus and striatum (Str). Two correlations (CpG 1 and 11) were individually significant prior to but not following correction, and this result could be expected by chance.
Correlations between hippocampus and blood (described in the text) yielded similar results, and no particular CpG yielded consistently high correlation across multiple tissues.”
The study focused on whether or not an individual’s experience-dependent oxytocin receptor gene DNA methylation in one of the four studied tissues could be used to infer a significant effect in the three other tissues. The main finding was NO, it couldn’t!
The researchers’ other findings may have been strengthened had they also examined causes for the observed effects. The “natural variation in maternal licking and grooming” developed from somewhere, didn’t it?
The subjects’ mothers were presumably available for the same tests as the subjects, but nothing was done with them. Investigating at least one earlier generation may have enabled etiologic associations of “the effects of early life rearing experience” and “individual variation in DNA methylation.”
https://www.sciencedirect.com/science/article/abs/pii/S0018506X1500118X “Natural variation in maternal care and cross-tissue patterns of oxytocin receptor gene methylation in rats” (not freely available)
A study of stress factors and neuroplasticity during infancy/early childhood
This 2015 French rodent study found:
“The coordinated actions of BDNF and glucocorticoids promote neuronal plasticity and that disruption in either pathway could set the stage for the development of stress-induced psychiatric diseases.
Genetic strategies that disrupted GR [glucocorticoid receptor] phosphorylation or TrkB [the BDNF receptor] signaling in vivo impaired the neuroplasticity to chronic stress and the effects of the antidepressant fluoxetine.
We demonstrate that fluoxetine prevented the neuroplasticity of chronic stress by priming GR phosphorylation at BDNF-sensitive sites.”
It wasn’t too difficult to see how many of the stressors had human equivalents during infancy/early childhood:
“To determine the plasticity of GR phosphorylation upon changes in the endogenous levels of BDNF and glucocorticoids, mice were exposed to a chronic unpredictable stress that included one daily random stressor for 10 consecutive days from P21 [immediately after weaning] to 1 mo of age.
Chronic unpredictable stress includes one of the following daily random stressors (wet bedding, no bedding, food deprivation, crowded cage, 2 h or 6 h restraining, forced swim, tail suspension).”
But who would give fluoxetine – Prozac – to a human infant or young child to prevent “the neuroplasticity of chronic stress” from having adverse effects?
http://www.pnas.org/content/112/51/15737.full “Neurotrophic-priming of glucocorticoid receptor signaling is essential for neuronal plasticity to stress and antidepressant treatment”
Inflexible behavior may be a byproduct of stress
This 2015 German human study found:
“15-mo-old infants exposed to stress thereafter kept performing a previously effective action, even after the action suddenly became ineffective.
Infants in a no-stress control group flexibly adjusted their behavior by disengaging from the newly ineffective action in favor of exploring an alternative action.
This finding demonstrates that stress impairs infants’ ability to adjust their behavior to changing circumstances.”
The primary measurement of stress levels was cortisol. Stressful conditions were:
- A stranger sat down next to them;
- A dancing robot played loud music and moved around;
- The infant’s caregivers left the room for up to four minutes.
News coverage stated that the study’s design was an adaptation of experiments that produced the same findings in adults. But would adult humans be stressed by being left alone for four minutes?
It’s likely that animal studies were the basis for some of this study’s experiments, as in the If research provides evidence for the causes of stress-related disorders, why only focus on treating the symptoms? study:
“Maternal separation in rodents is a useful model of early-life stress that results in enduring physiological and behavioral changes that persist into adulthood.”
A study limitation was that it involved just 26 infants.
http://www.pnas.org/content/112/41/12882.full “Stress impairs cognitive flexibility in infants”
Are a child’s genes the causes for their anxiety?
This 2015 Wisconsin macaque study was another attempt to justify the school’s continuing captivity of thousands of monkeys. The researchers performed a study that – if its experimental design was truly informative for helping humans – could have been done with humans.
A problem I saw in the news coverage was that the finding of:
“35 percent of variation in anxiety-like tendencies is explained by family history”
was attributed to genetics, with headlines such as “Anxious Brains Are Inherited, Study Finds.” The lead researcher encouraged this misinterpretation with statements such as:
“Over-activity of these three brain regions are inherited brain alterations that are directly linked to the later life risk to develop anxiety and depression.”
However, the researchers produced this finding by running numbers on family trees, not by studying genetic samples to assess the contributions of genetic and epigenetic factors!
The study’s “family history” correlation was different than finding an inherited genetic causation that wasn’t influenced by the subjects’ caged environments!
The study found:
“Metabolism within a tripartite prefrontal-limbic-midbrain circuit mediates some of the inborn risk for developing anxiety and depression.
The brain circuit that was genetically correlated with individual differences in early-life anxiety involved three survival-related brain regions. These regions were located in the brain stem, the most primitive part of the brain; the amygdala, the limbic brain fear center; and the prefrontal cortex, which is responsible for higher-level reasoning and is fully developed only in humans and their primate cousins.”
The 592 subjects were the human-equivalent ages of 3 to 12 years old. Primate brainstems and limbic systems are fully-developed BEFORE these ages.
The researchers skipped over potential evidence for the important contributions of epigenetic factors to “the later life risk to develop anxiety and depression” that change the studied brain areas during womb-life, infancy, and early childhood. Studies such as:
- Epigenetic changes in the developing brain change behavior
- Stress in early life can alter physiology and behavior across the entire lifespan
show:
- A developing fetus adapts to being constantly stressed by an anxious mother.
- When these adaptations persist after birth, they may present as physiological and behavioral maladaptations of the infant and young child to a non-stressful environment.
- Later in life, these enduring changes may be among the causes of symptoms such as the anxious overreactions the current study found.
http://www.pnas.org/content/112/29/9118.full “Intergenerational neural mediators of early-life anxious temperament”
Unconscious stimuli have a pervasive effect on our brain function and behavior
This 2015 Swedish human study, performed at the institution that awards the Nobel Prize in Physiology or Medicine, found:
“Pain responses can be shaped by learning that takes place outside conscious awareness.”
Images of neutral male faces were used as conditioning stimuli which the subjects were trained to associate with levels of pain.
The concluding sentence of the study:
“Our results demonstrate that conscious awareness of conditioned stimuli is not required during either acquisition or activation of conditioned analgesic and hyperalgesic responses, and that low levels of the brain’s hierarchical organization are susceptible for learning that affects higher-order cognitive processes.”
From the study’s abstract:
“Our results support the notion that nonconscious stimuli have a pervasive effect on human brain function and behavior and may affect learning of complex cognitive processes such as psychologically mediated analgesic and hyperalgesic responses.”
Principles of Dr. Arthur Janov’s Primal Therapy related to this study’s findings are:
- Experiences associated with pain can be remembered below our conscious awareness.
- Unconscious memories associated with pain, when activated, have varying forms of expression as they pass up through our levels of consciousness.
- These memories, when activated, have effects on our feelings, thinking, health, brain functioning, and behavior that are usually below our conscious awareness.
I’ll use one of Dr. Janov’s 2011 blog posts, On Being Alone, to show an example of how the study’s findings of:
- “Conscious awareness of conditioned stimuli is not required during either acquisition or activation of conditioned..responses” and
- “Nonconscious stimuli have a pervasive effect on human brain function and behavior”
are seen through the lens of Primal Therapy:
| Unconscious memories associated with the pain of being left alone may be stored, especially in the developing brain, in our lower brain areas below conscious awareness: | “Pain of being left alone a lot in childhood and infancy, added to the ultimate aloneness right after birth when no one was there for the newborn. That imprints a primal terror where a naïve, innocent and vulnerable baby has no one to lean on, to be held by, to snuggle up to, to be comforted. To be loved.” |
| As we develop, the cumulative memories associated with the pain of being left alone, when activated, may affect our feelings, thoughts, and behavior: | “And that also has multiple meanings: no one wants me; there is no one there for me: no one wants to be with me; I have no love and no one who cares. One races to phone others so as not to feel alone. One runs from the feeling and struggles mightily not to be alone. Or, depending on earlier events one stays alone out of that same feeling. These are by and large the depressives.” |
| Although memories associated with the pain of being left alone may be formed in our early lives, they remain decades later, and can be activated below our conscious awareness: | “When something in the present occurs which is similar to an old feeling “I am all alone and no one wants me,” the old feelings are triggered off..and the whole feeling rises toward conscious/awareness where it must be combated. Either the person wallows in the feeling and is overwhelmed by it even when she doesn’t even know what “it” is. Or the compounded feeling drives the act-out, forcing the person into some kind of social contact.” |
A PNAS commentary on the study stated:
“Pain, analgesia, and hyperalgesia represent higher-order cognitive functions.”
and attempted to draw conclusions from this reasoning.
The commentator was incorrect regarding pain. I didn’t see where this study showed or even postulated that pain was always a higher-order cognitive function. In fact, the researchers cited a sea slug study and stated:
“It would not be surprising if vestiges of simpler nonconscious processes would also be operative under some conditions.”
Maybe it would have provided clarifications if the researchers specifically defined “low” and “higher” used throughout the study in statements such as the closing sentence:
“Low levels of the brain’s hierarchical organization are susceptible for learning that affects higher-order cognitive processes.”
http://www.pnas.org/content/112/25/7863.full “Classical conditioning of analgesic and hyperalgesic pain responses without conscious awareness”
This post has somehow become a target for spammers, and I’ve disabled comments. Readers can comment on other posts and indicate that they want their comment to apply here, and I’ll re-enable comments.
