A 2025 rodent study investigated synergistic effects of sulforaphane (SFN) and nicotinamide mononucleotide (NMN) on diabetic cardiomyopathy:
“Diabetic cardiomyopathy (DCM) as a significant diabetes complication remains a major human challenge. In this study, we provide evidence that the fat mass and obesity-associated protein (FTO) plays a pivotal role in DCM pathogenesis.
Downregulation of FTO in DCM acts as a critical inducer of ferroptosis by increasing expression of acyl-CoA synthetase long-chain family 4 (ACSL4), a key positive mediator of ferroptosis. FTO-mediated mitigation of ferroptosis occurs in an ACSL4-dependent manner which leads to increased methylation of Acsl4 transcripts.
- Ferroptosis plays an essential role in the pathogenesis of DCM.
- As the most widespread mRNA modification, N6-methyladenosine (m6A) is globally downregulated and implicated in diabetes and its complications.
- FTO, which is an m6A demethylase, was found to be downregulated in diabetes and its cardiovascular complications.
- NAD+ enhances the demethylase activity of FTO. Dietary supplementation with NMN, a critical intermediate in the NAD+ biosynthetic pathway, has been shown to efficiently elevate endogenous NAD+ levels.
Enhancing the demethylase activity of FTO with NMN combined with SFN targeting NRF2 could synergistically reduce the level of lipid peroxides to inhibit ferroptosis, providing an effective avenue for alleviating DCM.
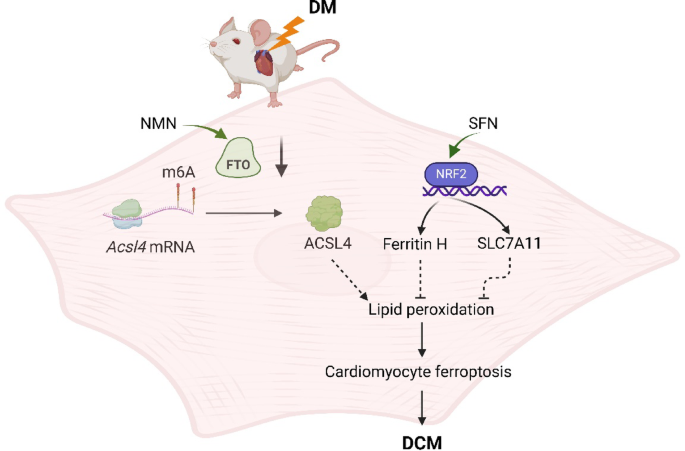
We found that NMN could alleviate ferroptosis and improve heart function through enhancing FTO. SFN could prevent ferroptosis and partly rescue heart function via AMPK-mediated NRF2 activation.
We demonstrated that SFN combined with NMN treatment could significantly inhibit lipid peroxidation and rescue cardiac function in DCM compared to SFN or NMN treatment alone.

Although the combined regimen further suppressed ferroptosis and improved cardiac performance, it fell short of complete remission, underscoring that additional pathways also contribute substantially to the pathogenesis of DCM.”
https://link.springer.com/article/10.1007/s12012-025-10080-w “FTO-Mediated Mitigation of Ferroptosis Occurs in an ACSL4-Dependent Manner in Diabetic Cardiomyopathy”
The epigenetic mechanism involved with this study’s dietary dissolved-in-water 100mM NMN dose was Non-CpG methylation. This study used the same very low sulforaphane dose intraperitoneally injected as Eat broccoli sprouts for your heart. Discussion of that study provided an example that if a person waited until a diabetes-related disease condition became a problem, capabilities to adequately address causes and prevent the problem may be lost.
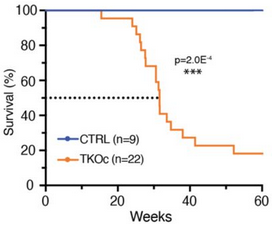
Notice in the last bar of the second graphic above taken from Figure 7 that the combined treatment was also provided to non-diabetic mice. These researchers provided over a dozen other measurements in Figure 7 to show similar short-term non-effects of the combined treatment, i.e. that it neither benefited nor harmed non-diabetic subjects. Grok interpreted this study’s 3-month-long intervention to be a 1-to-5 year human equivalent, depending on the measured effect (shorter for metabolic effects like MDA, longer for structural cardiac changes like reduced ferroptosis.)
The male subjects began at 2-months old, a human-equivalent 15-20 years old. These researchers gave them diabetes by feeding them a “high-fat diet for 3 months to induce insulin resistance, followed by a single intraperitoneal injection of streptozotocin (STZ) (in 0.1 mol/L of citrate acid buffer, 60 mg/kg) to induce partial insulin deficiency.” A 5-months old mouse is a 25-30 years old human equivalent.
Grok considered this study’s NMN human equivalent dose to be extremely high if provided in drinking water, but not if injected, depending on volume. However, the study didn’t state that its NMN dose was injected, and there was no dose volume indicated.