This 2021 study investigated environmentally-organized gut microbiome functional relationships:
“There has been a substantial gap between understanding microbiome assemblage and how its functionality is organized. In this study, we demonstrated the usefulness of metaproteomics in gaining a system-level understanding of microbiome functionality.
Our current finding highlights the value of further investigation into functional hubs and hub functions in microbiome proteomic content networks. This will provide a unique and systematic insight for prediction of community functional responses, or manipulation of microbiome functioning.
Across all metaproteomics datasets, Eubacterium, Faecalibacterium, Ruminococcus, Bacteroides, Clostridium and Coprococcus were found to be the most frequent functional hubs.
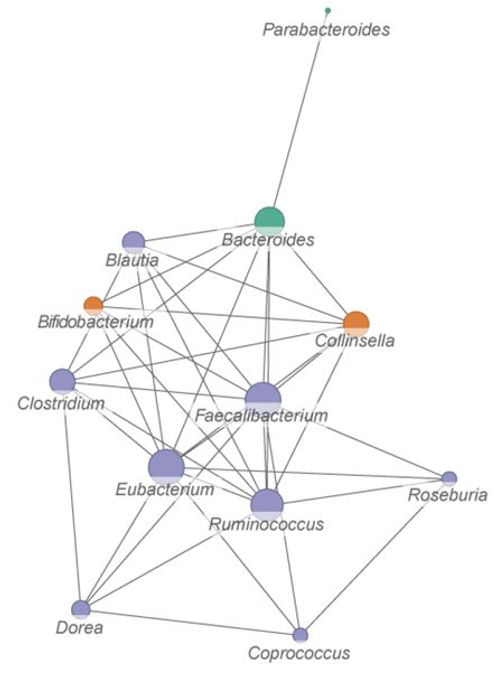
Taxon-function bipartite network based on functional distances between microbial genera. Size of a node corresponds to its degree.
Highly connected functions were enriched in metabolism of carbohydrates and amino acids, suggesting that microbial acquisition of nutrients from the environment and trophic interactions between microbes could be major factors that shape their active functional organization. Our result showing robustness of between-taxa functional distances across individual microbiomes implied a more fundamental mechanism that underlies selective organization of microbiome functionalities by environment.
We observed a universal pattern of between-taxa functional distances (dij) across all analyzed datasets. Notably, this pattern was fully shifted by a global increase in dij values, and subsequently a significant decrease of normalized taxonomic diversity in a subset of inflammatory bowel disease samples mostly obtained from inflamed areas.
This finding may support, from a functional angle, the hypothesis that there are alternative stable states (bi-stability or multi-stability) in the gut ecosystem. One frequently discussed mechanism behind these alternative states has been continuous exposure of the microbiome to a altered environmental parameter:
- An inflamed area in the gut will have a reduced mucus layer and elevated host defense responses.
- Host mucus layer is a nutritional source of cross-feeding in the gut microbiome.
- Loss of this layer may firstly affect network hub functions of carbohydrate and amino acid metabolism, and subsequently affect functional interactions in the whole community.
In addition, host defense responses attenuate microbial oxidative stress responses, which have been associated to microbiome dysfunction. Decrease of within-sample functional redundancy has been associated with impaired microbiome stability and resilience.
Resilient microbiota resist external pressures and return to their original state. A non-resilient microbiome is likely to shift its composition permanently and stay at an altered new state instead of restoring to its original state of equilibrium.”
https://www.biorxiv.org/content/10.1101/2021.07.15.452564v1.full “Revealing Protein-Level Functional Redundancy in the Human Gut Microbiome using Ultra-deep Metaproteomics”
My top genus Faecalibacterium – a cross-feeding, acetate-consuming, butyrate-producing commensal – would be more than twice the size of this study’s Faecalibacterium network projection in the above graphic. In this year’s efforts to make my gut microbiota happy, I’ve apparently done much to express its relevant gene network.

I came across this study by it citing Gut microbiota guilds.

